
Dissecting molecular mechanisms of immune microenvironment dysfunction in multiple myeloma and precursor conditions
 0
0 , ...
, ... Abstract
Multiple myeloma (MM) is a disease of clonally differentiated plasma cells. MM is almost always preceded by precursor conditions, monoclonal gammopathy of unknown significance (MGUS), and smoldering MM (SMM) through largely unknown molecular events. Genetic alterations of the malignant plasma cells play a critical role in patient clinical outcomes. Del(17p), t(4;14), and additional chromosomal alterations such as del(1p32), gain(1q) and MYC translocations are involved in active MM evolution. Interestingly, these genetic alterations appear strikingly similar in transformed plasma cell (PC) clones from MGUS, SMM, and MM stages. Recent studies show that effectors of the innate and adaptive immune response show marked dysfunction and skewing towards a tolerant environment that favors disease progression. The MM myeloid compartment is characterized by myeloid-derived suppressor cells (MDSCs), dendritic cells as well as M2-like phenotype macrophages that promote immune evasion. Major deregulations are found in the lymphoid compartment as well, with skewing towards immune tolerant Th17 and Treg and inhibition of CD8+ cytotoxic and CD4+ activated effector T cells. In summary, this review will provide an overview of the complex cross-talk between MM plasma cells and immune cells in the microenvironment and the molecular mechanisms promoting progression from precursor states to full-blown myeloma.
Keywords
INTRODUCTION
Multiple myeloma (MM) is a disease characterized by clonal expansion of terminally-differentiated plasma cells (PCs) in the bone marrow (BM)[1]. It is the second most common hematologic malignancy in the United States[2]. MM typically manifests clinically with end-organ damage consisting of anemia, renal impairment, lytic bone fractures, and hypercalcemia[1]. Over the past three decades, the introduction of novel treatments, such as proteasome inhibitors (PI), immunomodulatory drugs, autologous hematopoietic cell transplantation, and targeted monoclonal antibodies, has significantly improved the quality and length of life of patients with MM [Figure 1][3]. However, de novo resistance has been reported, and acquired resistance is almost inevitable over time, contributing to the incurable nature of this disease[4]. Disease refractoriness is driven by tumor intrinsic and extrinsic factors. MM is characterized by genetic heterogeneity of malignant PC clones and therapy-induced clonal evolution may play a significant role in disease progression[5-7]. Extensive research has demonstrated that extrinsic factors such as a permissive immune microenvironment influence tumor cell behavior and disease outcome.
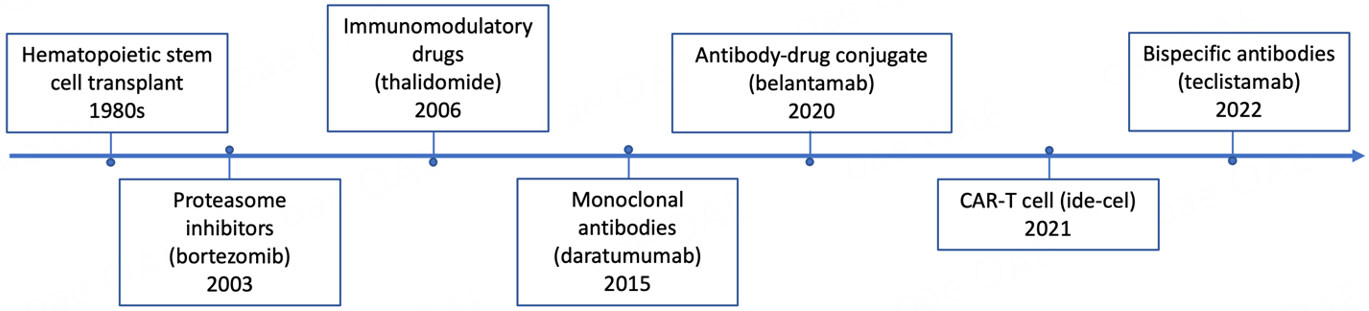
Figure 1. FDA-approved drugs for multiple myeloma and date of their first approval.
MM is almost always preceded by precursor conditions, monoclonal gammopathy of unknown significance (MGUS), and smoldering MM (SMM), through largely unknown molecular events[8,9]. Since premalignant states do not always progress to active myeloma, treatment is currently not justified solely on laboratory abnormalities, in the absence of symptoms. Genetic alterations of the malignant PC play a critical role in patient clinical outcomes. Del (17p), t(4;14), and additional chromosomal alterations such as del(1p32), gain (1q) and MYC translocations are involved in active MM evolution[10]. Interestingly, these genetic alterations appear strikingly similar in transformed PC clones from MGUS and SMM stages[11,12]. Emerging evidence indicates that tumors represent a complex ecosystem, and the combination of different conditions leads to a dynamic and self-fostering dysregulation of the immune system that supports tumor formation. Compositional and genetic expression changes of individual immune cell subtypes correlate with tumorigenesis and therapeutic outcomes[13].
This review will navigate the immune system dysregulation observed in MM, exploring the molecular mechanisms and the dynamic cross-talk between the tumor and the microenvironment that is responsible for skewing immune cells towards tolerance.
MYELOID COMPARTMENT
Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of myeloid cells in different stages of maturation. Previous studies have shown an increase in MDSCs in peripheral blood and BM of MM patients compared to healthy donors and MGUS patients[14,15]. In humans, MDSCs are commonly defined as the CD11b+CD33+HLADR-/lo population in the mononucleated cells[16]. There are two main subsets identified based on the additional expression of surface markers: CD15 for granulocytic MDSC
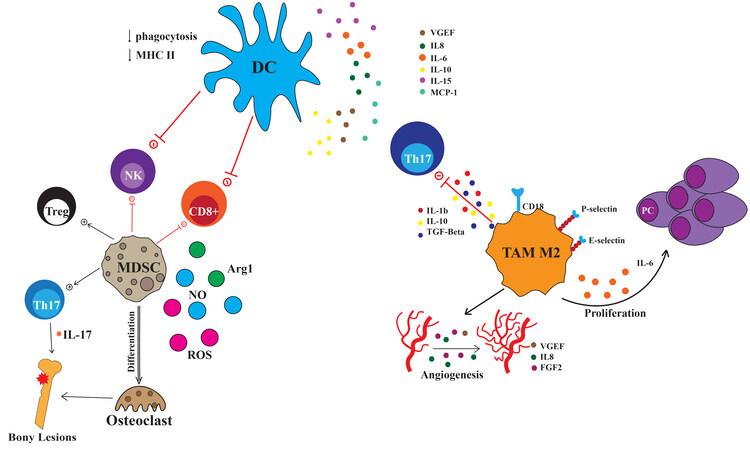
Figure 2. Myeloid cells in MM niche. Cartoon representing the cross-talk between myeloid derived suppressor cells (MDSC), dendritic cells (DC), Tumor-associated macrophages 2 (TAM M2) with MM plasma cells (PC) and Th17, T regulatory cells (Treg), Th17, cytotoxic CD8+ cells and natural killer cells (NK).
In addition to the immunosuppressive functions, across different tumor models, including MM, MDSCs serve as osteoclast progenitors, suggesting a role of these cells in cancer-associated lytic bone lesions[23,24]. Zoledronic acid, a commonly used bisphosphonate, inhibits osteoclastogenesis and concomitantly decreases MDSCs in the BM[23].
Daratumumab is a monoclonal antibody targeting CD38 and is now widely integrated into MM treatment combinations. Another mechanism of action through which this drug kills the myeloma cells is by creating an immunosuppressive environment through MDSCs depletion[25,26]. A recent study showed that CC motif chemokine ligand 5 (CCL5) and macrophage migration inhibitory factor (MIF) are molecules secreted by MM cells and have been found to be positive mediators of MDSC induction[27]. The same study found that IMiDs, such as lenalidomide and pomalidomide, decrease the secretion of CCL5 and MIF, resulting in suppressive effects on MDSC cells[27].
Neutrophils
Neutrophils are terminally differentiated cells that eliminate microbes and protect us against infections. However, cancer research on the role of tumor-associated neutrophils (TAN) has delivered controversial results[28]. Studies have shown that TANs possess both antitumor activities (e.g., direct cytotoxicity and inhibition of metastasis) and pro-tumor properties (e.g., promote angiogenesis, stimulate tumor cells migration and invasion, and support an immunosuppressive environment)[28]. Only in 2015, researchers found that neutrophils in cancer have a functional plasticity and can undergo “alternative activation” in response to signals from the tumor microenvironment. For example, the presence of transforming growth factor- TGF-) promote a pro-tumor phenotype (or N2 neutrophils), whereas interferon- INF) polarizes neutrophils towards an antitumor phenotype (or N1 neutrophils)[28]. Furthermore, multiple heterogeneous subsets have been observed. These subsets of neutrophils have the same immune phenotype but different patterns when undergoing density gradient centrifugation. Based on physical properties, these cells are now referred to as high-density neutrophils (HDNs) that sediment to the bottom, and low-density neutrophils (LDNs) that sediment at the top[29]. It is currently accepted that LDNs include g-MDSCs that have immunosuppressive properties, as described in the dedicated section. Additionally, the HDN subtype has been shown to have an N1-like phenotype and to kill tumor cells[28,29].
Neutrophils from peripheral blood from MM patients have a different gene expression profile compared to those isolated from healthy donors or MGUS[30]. Compared to both healthy donors and MGUS, neutrophils from MM patients expressed dysregulated genes in several biological processes, including endocytosis,
The neutrophil-to-lymphocyte ratio (NLR) has been introduced as a prognostic factor for survival and response to treatment in many tumor types. In MM, the NLR can predict the outcomes at diagnosis or after treatment with novel agents as well as autologous stem cell transplantation[30-32]. A recent single-cell RNA sequencing article reported that only the frequency of mature neutrophils at diagnosis (and not other granulocytic progenitors) is significantly associated with patient outcome[33]. The same study showed that a high ratio of mature neutrophils/T cells at diagnosis is correlated with inferior progression-free survival (PFS)[33].
Monocytes and macrophages
Monocytes and macrophages constitute a heterogenous multi-functional cell population. Circulating monocytes are recruited into the tumor microenvironment (TME), where they are converted to tumor-associated macrophages (TAMs)[34]. Activated TAMs are generally classified into two types: an immunoreactive antitumoral M1 phenotype (classically activated) and an immunosuppressive pro-tumoral M2 phenotype (alternately activated). Fully polarized M1 and M2 macrophages are the extremes of a continuum of functional states[35].
Macrophages heavily infiltrate the BM of myeloma patients relative to the BM of healthy controls[36,37]. Myeloma-associated macrophages (MAMs) in the BM niche are predominantly skewed phenotypically and functionally toward M2 phenotype[36,37]. MAMs provide nurturing signals to MM cells, promote immune escape, and negatively correlate with patient survival[38]. Interactions between integrins on MAMs and MM cells induce Src, Erk1/2 kinases, and c-Myc pathways, suppressing caspase activation and supporting tumor cell survival[39]. Besides contact mechanisms, human MAMs constitute a relevant source of pro-tumoral interleukin-1b IL-1), IL-10, IL-6, and tumor necrosis factor-alfa [Figure 2][40].
Importantly, macrophages also support MM progression through direct and indirect action on MM-associated neo-angiogenesis. Studies have found that angiogenesis is differently regulated in myeloma compared to precursor stages, suggesting that dysregulation in angiogenic cytokines and cells as well as hypoxia may contribute to myeloma progression[41-43]. Endothelial cells (EC) and epidermal growth factor receptor (EGFR) signaling have been shown to promote bone marrow angiogenesis and disease progression in preclinical models[42]. Interestingly, myeloma macrophages also secrete proangiogenic factors such as vascular endothelial growth factor (VEGF), IL-8, and fibroblast growth factor-2 (FGF-2)[44]. Macrophages derived from MM patients exposed to VEGF and FGF also show vasculogenic mimicry by acquiring endothelial cell markers and generating capillary-like vessels, in contrast with macrophages from normal subjects or MGUS[44]. Agents that block the VEGF signaling normalize the vasculature, improving oxygenation and delivery of chemotherapies to tumor cells while limiting the perfusion of the hyper-vascularized tumor areas. Clinical trials in MM tested anti-angiogenic agents such as bevacizumab used in combination with other MM drugs[45-47].
Several studies report that MAMs protect myeloma cells from chemotherapy-induced apoptosis, thereby contributing to melphalan and bortezomib resistance[37,48]. Recent studies show that the IKZF1 IRF4/IRF5 axis is relevant to drive M2 pro-tumoral skewing[49]. Interestingly, lenalidomide, which is an immunomodulatory drug commonly used in MM, induces cytotoxicity partially through inhibition of this axis[50].
Notably, therapeutic approaches aimed at depleting, inhibiting, or reprogramming macrophages have shown promising results in seminal preclinical cancer models. Currently, a common approach to induce macrophage depletion involves targeting the CSF-1 receptor (CSF1R), an important mediator of macrophage survival and differentiation. A recent study has demonstrated that anti-CSF1R antibody reduces tumor burden and improves survival in MM preclinical models[51]. In line with the unique plasticity of macrophages, MAMs retain their tumoricidal potential, making macrophage-repolarization an intriguing therapeutic strategy[52]. Current evidence from preclinical models shows that exposing MAMs to a cocktail of cytokines promoting M1 and contrasting M2 phenotype mediate M1-reprogramming[53]. An alternative strategy to achieve a similar result involves blocking the immune checkpoint CD47, a “don’t eat me” signal expressed on MM cells[54]. One study showed that Ruxolitinib, a Jak1/Jak2 inhibitor, induces an increase in the M1/M2 ratio in myeloma, suggesting that skewing towards a tumor-suppressive M1 phenotype is feasible[55].
Dendritic cells
Dendritic cells (DCs) are antigen-presenting cells (APCs) that play prominent roles in mediating both innate and adaptive immune responses[56]. DCs derive from BM mononuclear cells and are classified based on developmental stage into immature and mature DCs[57]. During the maturation process, DCs acquire HLA-DR expression, costimulatory molecules such as CD80, CD86 and generate specific T/B cell responses through the so-called cross-presentation process[58]. DC are further classified according to their origin, phenotype, and function in myeloid DCs (mDCs) and plasmacytoid DCs (pDCs). Evidence about the role of DCs in MM pathogenesis is controversial and the mechanism by which DCs contribute to the immunosuppressive microenvironment is not fully uncovered. pDCs interaction with MM cells stimulates the secretion of soluble factors, such as IL-10, VEGF, IL-8, IL-15, MCP-1, and IL-6, in the BM niche
Our group showed an increase in pDCs accumulation in the BM of MM patients compared to healthy donors[62]. We also reported that pDCs derived from the BM of MM patients have a decreased capacity to stimulate T cell response, support growth of MM, and contribute to drug resistance through secretion of cytokines such as SDF-1 (CXCL12) and IL-3[62].
However, the function of DCs in MM patients is still controversial. One group suggested that, based on the viability of MM cells, BM DCs play a dual and opposing role. Specifically, apoptotic malignant plasma cells undergo phagocytosis by bone marrow mDCs and pDCs, leading to the generation of tumor-specific cytotoxic T cells. However, the interaction of mDCs with nonapoptotic tumor plasma cells induces evasion from human leukocyte antigen (HLA) class I-mediated CD8 T cell killing by downregulating the synthesis of proteasome subunits in these cells and processing of antigens[64]. In this context, it has been shown that MM-derived pDCs and MDSCs express high levels of cell surface programmed-death-ligand 1 (PD-L1). The binding of PD-L1 to its receptor, programmed death-1 (PD-1), activates downstream signaling pathways and triggers apoptosis and anergy of T and NK effector cells, conferring a cancer immune-tolerant environment[62,65].
Genomic instability is an increased tendency to acquire genomic alterations and is one of the hallmarks of myeloma, both at early and advanced stages[66]. Interestingly, not only do the DCs support tumor growth, but they also regulate the genomic integrity of MM cells. Koduru et al. reported that the interaction between myeloma and DCs leads to rapid induction of the activation-induced cytidine deaminase (AID) enzyme and AID-dependent double-strand DNA breaks in myeloma cell lines as well as primary MM cells[67].
Further, DCs may be implicated in the development of osteolytic lesions in MM patients. It has been shown that DCs induce the expansion of polyfunctional Th17 in the BM, followed by IL-17 secretion, a potent pro-osteoclastogenic factor[68].
These data suggest that DCs may directly impact the biology of MM. Based on these premises, several drugs targeting DCs-MM interaction are under investigation. The inhibition of the CD28/CD80/CD86 axis with CTLA4 inhibitors is currently being explored in MM patients[68,69]. Additionally, pDCs express high levels of CD38 and daratumumab has been shown to cause depletion of pDCs[70].
LYMPHOID COMPARTMENT
Tregs and Th17
Naïve CD4+ T lymphocytes differentiate into T helper 1 (Th1), Th2, Th17, and regulatory T cells (Tregs) depending on the combination of cytokines in the microenvironment [Figure 3]. The complexity of T-cell immunity has been extensively investigated in myeloma and precursor diseases. Abnormalities in the function and distribution of T cell subsets have been reported in active MM, including expansion of Tregs and pro-inflammatory Th17, reduced Th1/Th2 ratio cytokine production, and altered stem-like capacity of the T cell compartment[68,71,72].
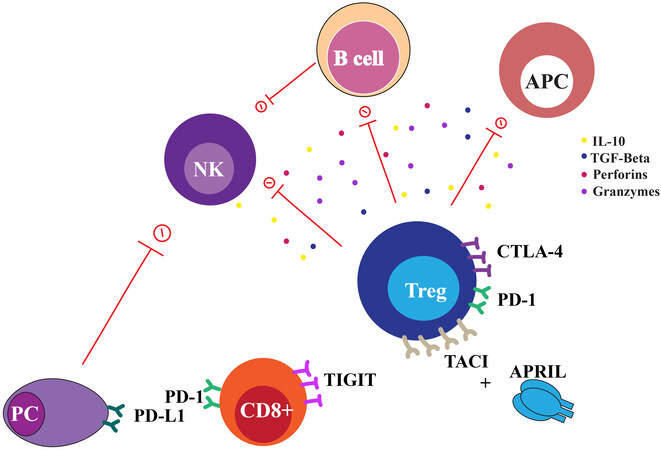
Figure 3. Lymphoid cells in multiple myeloma niche. Cartoon representing the interaction between lymphoid compartment and MM plasma cells (PC). Cytotoxic T cells CD8+ and regulatory T cells (Treg) in multiple myeloma have increased expression of immuno-suppression molecules, with suppression of B cells, APC and NK cells activity.
Tregs are a specialized subset of CD4+ T cells that have been associated with immune evasion in cancers. Tregs suppress the function of APCs, B cells, NK cells, and tumor-specific effector T cells by direct cellular interaction or by secretion of anti-inflammatory cytokines (e.g., IL-10 and TGF- β) and cytolytic granules (e.g., granzymes, perforins)[73,74]. An expansion of Tregs in the peripheral blood has a negative impact on survival and has been associated with a higher tumor burden in myeloma[72,75]. Recently, a preclinical study showed that in vivo depletion of Tregs in a MM murine model evokes a potent CD8+ T cell- and NK cell-mediated immune response, resulting in tumor regression[76].
Similar to pDCs and MDSCs, the PD-1/PD-L1 axis has also been studied in MM Tregs. Co-culture of CD4+ T cells with MM cells results in the generation of functional Tregs in a contact-dependent and antigen-presenting cell-independent manner. These Tregs show increased PD-1 expression compared to naturally occurring Tregs[77]. Preclinical studies have shown greater PD-L1 expression on myeloma plasma cells compared to MGUS or healthy donor plasma cells, contributing to immune escape mechanisms[78-80].
APRIL is a ligand for both BCMA and TACI (transmembrane activator calcium modulator and cyclophilin ligand interactor) receptors. Preclinical studies show that TACI is highly expressed in Tregs of MM patients and APRIL serum levels are increased in MM patients [Figure 3][81]. APRIL increases MM-driven Tregs via TACI-dependent proliferation associated with upregulation of immunosuppressive cytokines, such as IL-10, TGF β-1, and CD15s[81]. Additionally, APRIL binds BCMA receptor and is highly secreted by MM-derived osteoclasts, potentially contributing to myeloma bone disease[82].
The therapeutic implications of Tregs are numerous. The immunomodulatory activity of lenalidomide is partly driven by the downregulation of inducible T-cell costimulatory ligands, a decrease in Treg population, and their respective FoxP3 expression[83,84]. Recently, a novel subpopulation of CD38-positive Tregs was identified. In vitro studies have shown that this subset of Tregs has a more potent immunosuppressive activity compared to CD38 negative Tregs and is significantly reduced in daratumumab-treated patients[25].
The Treg/Th17 balance in the microenvironment of MM patients is considered to be a marker of immunoregulatory control[85]. Physiologically, Th17 are pro-inflammatory cells and secrete among others, IL-17, IL-6, IL-22, and TNF-α cytokines. Seminal preclinical studies provide evidence that supports the pivotal role of Th17 and IL-17 in myeloma development and progression. MM patients show a significant imbalance in Treg/Th17 ratio when compared to either healthy donors or other monoclonal gammopathies and this correlates with worse long-term survival[86-88]. Clinically, the proportion of Th17 cells in the bone marrow positively correlates with tumor stage, serum lactate dehydrogenase, and serum creatinine concentration[87]. Preclinical studies reported an association between Th17 and MM cell proliferation, migration, neoangiogenesis, immune evasion, and myeloma bone disease[68,89]. Consistently, Noonan et al. showed that Th17 are enriched in the BM, where they mediate the development of lytic bone lesions via secretion of IL-17[88].
Cytotoxic T cells
CD8+ T cells are effector lymphocytes characterized by cytotoxic tumor-specific activity. Interestingly, CD8+ T cells are equally prominent in precursor conditions, active myeloma, and healthy donors. However, in MM, BM cytotoxic T cells have an altered capability to respond to tumor-specific antigens. Several mechanisms have been proposed to corroborate this immune evasion, including the ineffective antigen presentation capacity of the dendritic cells and a protective myeloid compartment[33,90-92]. Supporting this rationale, a preclinical study showed that T cells from patients with clinically progressive myeloma were found to induce a potent cytolytic activity against freshly isolated autologous tumor cells, only after ex vivo stimulation with autologous dendritic cells[93]. Subsequent studies showed that dendritic cells primed with myeloma cell lysates induce a potent tumor-specific cytotoxic T cell response[94]. These data provide evidence that endogenous cytotoxic T cells have the potential to be activated to elicit an anti-MM response.
MM and tumor microenvironment cells secrete IL-10, TGF-β, immunosuppressive ectoenzymes and other soluble factors, which potentially modulate the cytotoxic activity of CD8+ cells[95]. The ectoenzyme family relies on adenosine, which is a well-characterized immunosuppressive metabolite[96]. In the extracellular space, ATP is metabolized to adenosine by the sequential activity of CD39 and CD73, which are two extracellular enzymes. Specifically, CD39 converts ATP and ADP to AMP, and CD73 rapidly metabolizes AMP to adenosine[96,97]. MM cells express high levels of CD38 and CD39 surface molecules. Consistently, higher levels of adenosine are detected in the serum of patients with active myeloma compared to patients with precursor myeloma disorders and healthy donors[98-100]. Similarly, a recent study using a murine model of MM showed that inhibitors of the adenosine pathway induce activation of immune cells, increase interferon-gamma production and reduce myeloma tumor load[99].
Studies have shown that active myeloma is characterized by a dynamic alteration of CD8+ phenotype ranging from “senescent” to “exhausted”. CD8+ T cells express molecules associated with T cell exhaustion (PD-1, CTLA-4, CD160, 2B4, LAG3) and T cell senescence (CD57, KLRG-1, lack of CD28) [Figure 3]. Unfortunately, recent trials showed disappointing clinical benefits of anti-PD-1 blockade therapy in myeloma[101,102]. Importantly, TIGIT (T cell Immunoreceptor with immunoglobulin and ITIM domains) has recently emerged as a promising immune checkpoint in MM. High levels of TIGIT expression on CD8+
NK cells
NK cells are small granular lymphoid cells exerting cytotoxic activity against tumor cells. While studies reported an increase in NK cells in the peripheral blood and BM of MM patients, other studies revealed a decrease in this population[106-108]. Importantly, NK activity is impaired, especially in cases of clinically advanced MM disease[109,110]. To discriminate between target and healthy cells, NK cells express surface receptors that induce cytotoxic activation (such as NKG2D, NCR, DNAM-1, CD16) or inhibition (such as KIR, CD94/NKG2A)[111]. MICA is a well-known ligand present on tumor cells that binds the receptor NKG2D on NK cells. Studies have shown that as disease progresses, MICA is shed from the surface of MM cells and NKG2D is internalized, impairing NK cell activation and killing of the tumor cell[112-114]. Similarly, DNAM-1 expression is reduced as MM progresses while its ligand PVR is upregulated[114]. Interestingly, IL-6 has been associated with down-regulation of perforin expression through NF-kB and STAT3 pathways, potentially contributing to impaired NK cell cytotoxicity[115,116]. A decrease in NK cell surveillance and cytotoxicity against MM might also be partially driven by the up-regulation in the expression of PD-1 on NK cell surface, which accompanies the increase in PD-L1 expression on MM cells[117]. However, inhibitors of the PD-1/PD-L1 pathway have been ineffective as single agents in MM[118].
In patients with long-term disease, autologous stem cell transplantation induces an increase in NK population. Recent clinical trials have focused on boosting NK-related immunosurveillance via activation and expansion of NK cells ex vivo or by using allogeneic cord blood-derived NK cells[119,120]. A strategy to activate and expand functional NK cells is based on using engineered cells expressing ligands that induce NK activation combined with a cocktail of specific cytokines. Based on these biological premises, a recent seminal protocol for cytokine-induced memory-like (CIML) NK cell development was established[121]. Specifically, NK cells undergo ex vivo pre-activation with IL-12, IL-15, and IL-18 before administration to patients[121]. Our group is currently conducting a clinical trial employing the use of CIML NK cells along with low-dose IL-2 in newly diagnosed MM patients (NCT04634435)[121].
INFLAMMATORY MESENCHYMAL STROMAL CELLS
Mounting evidence from preclinical models reproducing MM cells in the microenvironment niche suggests that mesenchymal stromal cells (MSCs) support MM development and induce drug resistance and immunomodulation via direct cellular interaction and soluble factors[122,123]. A recent single-cell RNA sequencing study comprehensibly characterized an inflammatory phenotype of MSCs (iMSCs) nearly exclusive to the MM microenvironment[124]. The investigators proposed a model whereby soluble factors such as IL-1 secreted by monocytes and TNF secreted by NK and CD8+ lymphocytes promote the inflammatory phenotype in stromal cells. They also speculated that tumor cells present in the BM induce inflammatory MSC phenotype by activation of immune cells leading to the production of inflammatory cytokines and by releasing exosomes containing DAMPs.
In turn, iMSC secrete IL-6, LIF, and CCL2 that support tumor cell proliferation and IL-6, C3, ANXA-1, and VEGFa that modulate immune cell compartment, particularly myeloid cells [Figure 4A][124].
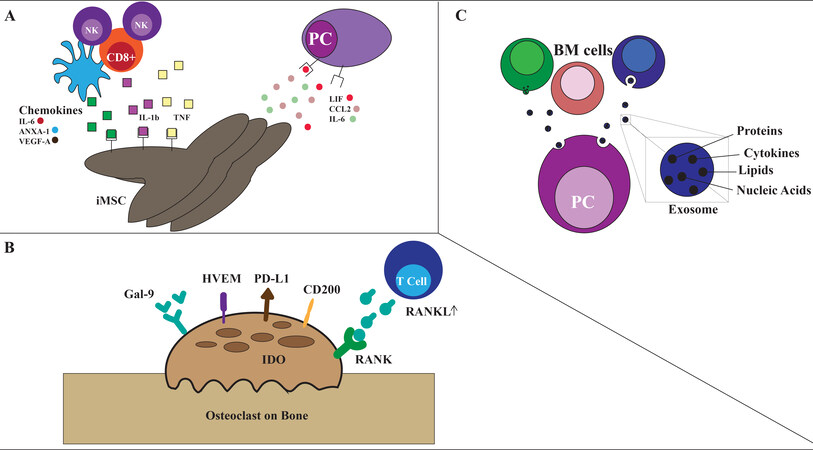
Figure 4. Other microenvironment components. (A) Inflammatory mesenchymal stromal cells (iMSCs): IL-1, TNF-alfa secreted by myeloid cells, cytotoxic T cells and NK cells and DAMPs deriving from tumor cells induce inflammatory phenotype of stromal cells. In turn, iMSC support tumor development through secretion of IL-6, LIF, CCL2 among other cytokines. iMSC simultaneously recruit and modulate immune cells, primarily myeloid cells. (B) osteoclasts: upregulation of Gal-9, CD200, HVEM, CD200, PD-L1, RANK in osteoclasts with secretion of IDO induce immuno-suppression; osteoclasts also produce OPN, IL-6, BAFF and APRIL that support MM survival. (C) Exosomes: BM cells in MM compared to healthy donors showed lower levels of miRNA-15a and increased levels of pro-tumoral Il-6, fibronectin, and CCL-2.
OSTEOCLASTS AS IMMUNOCOMPETENT CELLS
The imbalance between bone deposition and bone resorption is responsible for osteolytic bone lesions, a hallmark of myeloma development. Besides their function on bone metabolism, osteoclasts (OCLs) have an immunosuppressive and pro-tumoral role in the MM BM microenvironment. Preclinical studies show that OCLs produce MM pro-survival factors, such as osteopontin (OPN), IL-6, BAFF, and APRIL
OCLs inhibit T cells, which in turn enhance osteoclastogenesis. In fact, in MM co-culture systems, activated T lymphocytes secrete high levels of RANKL, the main pro-osteoclastogenic factor[127]. Consistently, MM patients with osteolytic lesions show RANKL up-regulation by BM T cells as compared to MM patients without bone lesions[127]. Lastly, MM T cells secrete IL-3 that promotes MM-induced osteoclastogenesis and levels of this cytokine are higher in MM patients compared to controls[128].
As described in previous paragraphs, MM patients show an increase in IL-17-producing Th17 that inhibits cytotoxic T-cell activity and promotes MM cell growth[68]. Interestingly, levels of cytokines that selectively induce Th17 phenotype tightly correlate with lytic bone lesions[68,88].
EXOSOMES
Exosomes are 30-100 nm small, secreted vesicles containing nucleic acids, proteins, and lipids. They are generated via multivesicular endosomes and are subsequently released upon fusion of these endosomes with the cell membrane [Figure 4C][129-131]. Recruitment and clustering of macromolecules generally occur either via endosomal sorting complex required for transport (ESCRT)-dependent or ESCRT-independent mechanisms[131,132]. Once secreted, exosomes serve as vital cross-talking mediators between the bone marrow microenvironment and surrounding MM[133,134]. In doing so, they promote angiogenesis, osteolysis, and drug resistance, contributing to MM progression[135]. The interaction of exosomes with surrounding cells occurs through distinct mechanisms, primarily mediated by the direct fusion with the plasma membrane, or incorporation via pathways such as phagocytosis. Upon release of exosome content, downstream intracellular signaling is subsequently activated[131]. Exosomes derived from MM cells have the capacity to reprogram cells in the bone marrow to promote a pro-tumor environment that supports disease progression[136]. This can be attributed to elements such as cell recruitment, immunosuppressive effects, and horizontal transfer of genetic information[133,134]. In vitro studies have demonstrated that exosomes derived from the bone marrow stromal cells (BMSCs) induce MM growth, survival, and drug resistance and subsequent disease progression[137]. Preclinical studies showed that exosomes secreted by BMSCs derived from MM patients promote tumor growth in contrast to exosomes derived from healthy patients that have demonstrated opposing effects[138,139].
In-depth profiling of the BMSCs exosomes content in MM compared to healthy donors, demonstrated lower levels of the tumor-suppressive factor miRNA-15a, and higher levels of pro-tumoral molecules such as chemokine C-C motif ligand (CCL) 2, IL-6, and fibronectin [Figure 4C][139].
Exosomes can also induce drug resistance mechanisms via inter-cellular transfer of molecules. In fact, it has been shown that exosomes containing specific molecules such as PSMA3 and PSMA3 Antisense RNA1 were transferred from PI-resistant MM patients to sensitive MM patients, inducing proteasome inhibitor resistance by increasing the proteasome activity[140].
Adding to the complexity of the system, the interaction between micro-environmental cells and MM cells dictates the composition of the exosomes in the ecosystem. Our recent work showed that co-culture of MM with BMSCs cells induces HDAC3 expression in BMSC cells, while HDAC3 knockdown in BMSC leads to quantitative and qualitative changes in secreted exosomes that ultimately contribute to MM cell growth arrest[141].
Within the bone marrow microenvironment, exosomes contribute to a pro-osteoclast microenvironment through non-coding RNAs (ncRNAs)[142,143]. Generally, high osteoclasts to osteoblasts ratio induces bone reabsorption and myeloma bone disease. Certain pathways have demonstrated increased osteoclastogenesis. Seminal studies have shown that molecules enriched in MM-exosomes such as lncRNA RUNX2 antisense RNA 1 (RUNX2-AS1), amphiregulin, and miR-129-5p increase osteoclastogenesis by reducing RUNX2 splicing efficiency, activating the epidermal EGFR pathway, and downregulating the expression of the transcription factor Sp1, respectively[144-146]. Exosomes have also shown the ability to downregulate osteoblastogenesis through the suppression of osteoblastic differentiation proteins such as Runt-related transcription factor 2 (Runx2), Osterix, and osteocalcin[145-147].
IMMUNE MICROENVIRONMENT MODULATION IN PROGRESSION FROM PRECURSOR STAGES TO ACTIVE MYELOMA
In recent years, genomic studies provided the opportunity to dissect the genetic alterations that occur in active myeloma and precursor stages with unprecedented accuracy. It has been shown that MGUS/SMM patients may already harbor chromosomal alterations that define MM. However, no driver genetic mutation has been identified to date; hence, the cause of MM pathogenesis and progression from MGUS/SMM/MM remains elusive. Importantly, dissecting the mechanisms of evolution of the immune microenvironment from precursor non-malignant stages to active myeloma could pave the way to develop strategies for immune-based patient stratification and therapeutic strategies aiming at delaying this progression and potentially eradicating MM.
A recent single-cell study investigating the cellular composition of the tumor microenvironment reported a significant, although heterogenous, enrichment of T cells, CD16+ monocytes, and NK cells at the MGUS stage[13]. It has been shown that mature CD14+ monocytes are already dysfunctional at the MGUS stage, presenting a phenotypic shift leading to loss of major histocompatibility complex class II (MHC II) expression. Therefore, CD14+ monocytes have an impairment of their antigen-presenting cell capacity with suppression of the T cell activation, as early as in the MGUS stage[13]. Studies on matched samples showed an expansion of monocytes and macrophages during the progression from SMM to full-blown MM[148]. Similarly, Calcinotto et al. showed that patients with active MM, compared to MGUS/SMM, present an expansion of macrophages in the BM microenvironment that associates with increased BM vascularity and poor prognosis[149]. The same group suggested that the progression from MGUS/SMM to MM is also driven by an “angiogenic switch” characterized by an increase in BM plasma levels of angiogenic cytokines[149]. This was further confirmed by a large prospective study that showed that a composite angiogenesis biomarker score, calculated based on the levels of EGF, HGF, and Ang-2, correlated with an increased risk of MGUS progression to MM[150].
In the context of APC cells, although researchers have speculated that the clinical progression from MGUS to MM may be driven by defects in the dendritic cell function, evidence supporting this assumption is controversial. While certain studies reported that mDCs and pDCs accumulate in the BM during MGUS to MM progression, others have shown a significant depletion of both circulating and BM pDCs in patients with MGUS and active MM, compared to healthy donors[64,151].
Seminal work has been done to extensively characterize the T-cell compartment and its potential role in myeloma development from precursor stages. Ex vivo T-cells derived from BM of patients with preneoplastic gammopathy retain a vigorous antitumor activity against premalignant plasma cells[152]. This is in contrast to T cells from myeloma bone marrow, which lack tumor-specific rapid effector function, suggesting that T cells in MM lose the ability to naturally control tumor progression[152]. Studies on the Vk*MYC MM mice model showed an accumulation of CD3+ T cells, both CD8+ and CD4+ T cells, during disease progression[149]. Analysis of the cytokine composition of CD4+ T cells revealed a progressive loss of Th1 immune response and skewing toward Th2 response in Vk*MYC compared to WT mice[149]. Conversely, a recent single-cell study showed a depletion of CD4+ lymphocytes and a heterogenous pattern of expression of CD8+ cells during the progression from SMM to MM in matched samples[148].
Among the immune system dysfunctions identified during the progression to MM, it is worthwhile mentioning the increased immunosuppressive T phenotype starting from the MGUS stage. Studies reported an expansion of immunosuppressive Tregs and T-cell exhaustion phenotype, starting from MGUS, suggesting that T-cell dysfunction might be an early event[13,72]. Th17 cells and gut microbiota might also play a role in the progression. A recent study on Vk*MYC mice showed that gut microbiota promote the differentiation into Th17 cells, which migrate to the BM, where they favor the progression from SMM to MM[153].
Lastly, changes in the NK cell compartment in the BM microenvironment have been reported in the progression of MGUS to MM[154]. In a recent single-cell study of matched samples, investigators have reported a highly dynamic microenvironmental NK profile over patients’ disease course, resulting in the arduous interpretation of a common trend for how tumor microenvironment evolves during disease progression[148]. Liu et al. reported that in the NK population, CXCR4-expressing NK was prevalent during active disease, while CX3CR1-expressing NK was more intensively represented post-transplant[148]. Similarly, another single-cell study by Zavidij et al. showed that in patients with high NK-cell infiltration, this fraction was predominantly constituted of CXCR4+ cells, while patients with fewer NK cells showed a shift toward CX3CR1 expression[13]. Importantly, previous studies have shown that these are chemoattractant receptors responsible for mediating homing to the BM[155,156]. This may explain the heterogenous representation of NK subsets observed in these studies and could suggest an MM-orchestrated mechanism of immune evasion.
IMMUNOTHERAPY IN MULTIPLE MYELOMA
Immunotherapy is a type of cancer treatment that boosts the immune system to recognize and kill tumor cells. In the past few decades, the understanding of the immune system composition and the better characterization of myeloma antigens have been instrumental in developing immunotherapies for the treatment of MM. Immune-based treatment approaches are gaining supremacy over traditional therapies not only in myeloma but also in other liquid and solid tumors. This can be explained by high efficiency and specificity, with a more manageable toxicity profile. Novel immunotherapies, such as CAR T cell therapy (CAR) and bispecific T cell engagers (BiTEs), are developed targeting different surface antigens, such as BCMA, SLAM7, CD38, or GPRC5D. Early use of immunotherapy may improve outcomes and several immunotherapy combinations have been recently approved for MM, and many others are under active investigation.
Chimeric antigen receptor T (CAR-T)-cell therapy
Chimeric antigen receptor T (CAR-T) cells are engineered T cells that express a specific antigen (Ag) TCR that allows recognition of tumor Ag and killing of the cell. In CAR-T therapy, the patients’ T cells are selected from the peripheral blood, edited to express the chimeric antigen receptor, expanded, and reinfused into the patient[122]. The rapid activation and expansion of T cells, upon binding to target tumor Ag, can potentially lead to life-threatening complications such as cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS)[157]. There are currently two FDA-approved CARs, both targeting BCMA that is highly expressed on malignant PCs: idecabtagene vicleucel (ide-cel/bb2121) and ciltacabtagene autoleucel (cilta-cel). A list of CAR T cell trials is summarized in Table 1. Ide-cel was the first CAR-T cell approved for patients with relapsed or refractory MM (RRMM), following the phase II KARMMA-1 trial results, showing an overall response rate (ORR) of 73%, median progression-free survival (PFS) of 8.6 months with 33% of patients achieving complete remission (CR)[158,159]. Recently, results from the phase 3 KARMMA-3 trial that recruited patients earlier in the disease course, after two to four previous lines, showed that at a median follow-up of 18.6 months, ORR was 71%, median PFS was 13.3 months, with overall survival data still immature[160]. Ide-cel is being tested in phase 1 KARMMA-4 trial in newly diagnosed multiple myeloma patients with high risk (R-ISS stage III), with the rationale that upfront use, where there may be more bone marrow reserve and less “exhausted” immune system, may offer an opportunity to replace transplant with CAR-T cell therapy. Cilta-cel was approved by the FDA for RRMM in February 2022 based on findings from phase I/II CARTITUDE-1 study, showing ORR of 97%, CR achieved in 82%, and 27-month PFS and OS rates of 55% and 70%, respectively[161]. Similar to ide-cel, cilta-cel is also being tested upfront in phase 3 CARTITUDE-5 trial for NDMM who are non-transplant-eligible (NTE) and in phase 3 CARTITUDE-6 in NDMM who are transplant-eligible [Table 1].
Summary of CAR-T cell clinical trials in MM
| Target | Trial name (name of the drug) - phase | Study population | Outcomes | NCT |
| BCMA | KARMMA1 (idecel) - phase 2 | RRMM | ORR 73%; CR 33%; mPFS 12.1 months - FDA approved | NCT03361748 |
| BCMA | KARMMA3 - Phase 3 | RRMM | ORR 71%; mPFS 13.3 months - FDA approved | NCT03651128 |
| BCMA | KARMMA4 - Phase 1 | High risk NDMM | NA | NCT04196491 |
| BCMA | CARTITUDE1 (ciltacel) - Phase 1b/2 | RRMM | ORR 97%; CR 82% - FDA approved | NCT04133636 |
| BCMA | CARTITUDE5 - Phase 3 | NTE NDMM | NA | NCT04923893 |
| BCMA | CARTITUDE6 - Phase 3 | TE NDMM | NA | NCT05257083 |
Despite the high response rates, the majority of patients with MM exposed to CAR T targeting BCMA relapse within two years[162]. Several mechanisms of CAR-T cell resistance were speculated: (1) intra-tumoral BCMA expression heterogeneity leading to the selection of low-BCMA expressing clones; (2) T cell exhaustion; (3) cleavage of BCMA by gamma-secretase enzymes, which releases a soluble BCMA (sBCMA), acting as a decoy; and (4) activation-induced cell death (AICD) of the T cells[163-165].
Bispecific T-cell engagers (BiTEs)
Bispecific antibodies (BsAbs) are antibodies with two binding sites that recognize two antigens. Two types of constructs exist immunoglobulin (IgG)-like with an Fc fragment and non-Ig-like that lack the Fc fragment. Bispecific T-cell engagers (BiTEs) are a non-Ig-like subtype of BsAbs, consisting of two antigen recognition domains, one for CD3 of the TCR complex on T cells and one for the specific tumor antigen on the cancer cell. Through this interaction, BiTEs redirect autologous T cells in proximity to the tumor cells to facilitate T-cell activation, cytokine release, and killing of the cancer cell[166]. Unlike CAR-T cells, BiTEs are antibody-based molecules that are available off-the-shelf, which allows faster delivery as a treatment. The adverse events are similar to CAR-T cells, as they both activate the immune system leading to CRS and neurotoxicity, though less extensively[167]. The main target for MM BiTE studies is BCMA, with few studies investigating CD38, CD19, GPRC5D, and FcRH5. Table 2 summarizes trials for BiTEs. In October 2022, teclistamab (CD3 × BCMA) was approved for heavily treated RRMM, who have received at least 3 prior lines, following the results of phase I/II MajesTEC-1 trial that showed ORR 63%, 39% achieving CR, median PFS of 11 months. The most common toxicities were CRS (72%), neutropenia (71%), and infections
Summary of BiTEs clinical trials in MM
| Target | Trial name (name of the drug) - phase | Study population | Outcomes | NCT |
| BCMA × CD3 | MajesTEC-1 (teclistamab) - phase 1/2 | RRMM | ORR 63%; CR 39%; mPFS 11 months - FDA approved | NCT04557098 |
| BCMA × CD3 | MajesTEC-9 (teclistamab) - phase 1/2 | RRMM | NA | NCT05572515 |
| BCMA × CD3 | MagnetisMM-1 (elratamab) - Phase 1 | RRMM | ORR 64%; CR 31% | NCT03269136 |
| CD3 × GPRC5D | MonumenTAL-1 trial, (talquetamab) - phase 1 | RRMM | ORR 70% | NCT03399799 |
| CD3 × FcRH5 | Cevostamab | RRMM | ORR 54% | NCT03275103 |
| CD3 × CD38 | GBR 1342 | RRMM | NA | NCT03309111 |
Elranatamab is another anti-BCMA BsAb under investigation. In phase 1 MagnetisMM-1 trial, heavily pre-treated RRMM received weekly subcutaneous elranatamab administration either alone, with lenalidomide or with pomalidomide. The ORR at the recommended phase 2 dose was 83%[169]. The updated follow-up data reported an ORR of 64%, with 31% of patients achieving CR. CRS was observed in 67% of patients with no grade 3 or higher CRS[170]. Overall, considering response rates and safety profile, elranatanab was granted breakthrough therapy designation by the FDA for the treatment of RRMM patients who have previously been treated with at least 4 prior lines of therapy[170].
Characterization of the myeloma cancer cells has been instrumental in guiding the design of CARs. BiTEs targeting other MM surface antigens other than BCMA are being developed and have demonstrated promising results. G-protein coupled receptor family C group 5 member D (GPRC5D) is a recently identified molecule highly expressed on malignant MM plasma cells in the BM, but not on other healthy cells. In phase 1 MonumenTAL-1 trial, Talquetamab (CD3 × GPRC5D) was administered subcutaneously weekly, reaching ORR of 70% and the FDA granted it a breakthrough therapy designation in July 2022 for the treatment of RRMM who have received at least 4 prior lines[171,172].
FcRH5 is a type 1 membrane protein that is almost uniquely expressed on B cells and plasma cells. Cevostamab (CD3 × FcRH5) has shown encouraging results in phase I study enrolling RRMM with an ORR of 54% at the 160 mg dose level in the expansion cohort.
CD38 is well known to the myeloma community as a surface molecule highly expressed in MM cells[100]. The clinical benefits of anti-CD38 mAb daratumumab led to studies engineering BiTEs targeting CD38.
As described above, numerous BiTEs trials are currently active and showing high response rates, with preliminary reports showing that BiTEs might be able to rescue the progression of disease in patients with advanced MM. The challenge that clinicians are encountering in real practice is (1) prevention of serious infections that can be deadly; (2) development of guidelines that direct clinicians on when to interrupt treatment with BiTEs; and (3) evaluating the safety of combination with other myeloma drugs.
Antibody-drug conjugates (ADC)
Antibody-drug conjugates (ADCs) are monoclonal antibodies that are conjugated with highly cytotoxic compounds via a chemical linker. ADC binding to a tumor-associated antigen induces internalization of the cytotoxic agent and tumor cell death, resulting in theoretically diminished toxicity against normal cells. Belantamab mafodotin is the first-in-class ADC designed to bind to BCMA on plasma cells. The FDA approved belantamab for RRMM who had been previously heavily treated with at least four lines. Approval was granted following the results of the phase II DREAMM-2 trial, which showed an ORR of 31%, median PFS of 2.8 months, and median OS of 13.7 months[174].
Several DREAMM studies are ongoing, as described in Table 3. Notably, the use of belantamab has resulted in frequent ophthalmologic toxicities such as corneal keratopathy (72%)[174].
Summary of antibody-drug conjugates (ADC) clinical trials in MM
| Target | Trial name (name of the drug) - phase | Study population | Outcomes | NCT |
| BCMA | DREAMM-2 (belantamab) - phase 2 | RRMM | ORR 31%; mPFS 2.8 months, mOS 13.7 moths - FDA approved | NCT03525678 |
| BCMA | DREAMM-3 (belantamab) - phase 3 | RRMM | NA | NCT04162210 |
| BCMA | DREAMM-8 (belantamab) - phase 3 | RRMM | NA | NCT04484623 |
| BCMA | DREAMM-9 (belantamab) - phase 1 | NDMM | NA | NCT04091126 |
Clinical trials investigating ADCs targeting other antigens and utilizing other cytotoxic agents are currently ongoing. These include lorvotuzumab mertansine (anti-CD56 conjugated to cytotoxic maytansinoid) and indatuximab ravtansine (anti-CD138 conjugated to cytotoxic maytansinoid)[165,175,176].
Monoclonal antibodies (mAbs)
CD38 MAbs
CD38 is a glycoprotein that is highly expressed on malignant PCs but is also present at lower levels on normal PCs, myeloid and lymphoid cells, red blood cells, and platelets[100]. Because CD38 is not exclusive to MM cells, targeting CD38-positive cells can induce off-target NK depletion as well as favorable depletion of immunosuppressive Tregs and Bregs[25,177,178]. Daratumumab (Dara) was the first mAb approved by the FDA for the treatment of MM, initially only for RRMM patients and in recent years also for NDMM, both transplant-ineligible and transplant-eligible. The results of the main anti-CD38 trials are summarized in Table 4. Daratumumab was first approved as monotherapy on November 16, 2015, based on the results of the SIRIUS trial that enrolled MM patients who have received at least three prior lines of therapy (median PFS 3.7 months, ORR 29%)[179]. In the following years, trials were designed to combine daratumumab with PIs bortezomib/velcade (V) and carfilzomib/kyprolis (K) or IMiDs thalidomide (T), lenalidomide/revlimid (R) and pomalidomide (P) with the addition of steroids dexamethasone (d) or prednisone (P). In 2016, daratumumab was approved for RRMM who have received at least one prior line, following results of the CASTOR trial (Dara-Vd; median PFS of 16.7 months) and the POLLUX trial (Dara-Rd; median PFS of 44.5 months)[180,181]. Importantly especially for older patients, in May 2018, based on an ALCYONE trial, Dara was approved for non-transplant-eligible NDMM in combination with bortezomib, melphalan, and prednisone (Dara-VMP; median PFS of 36.4 months)[182]. Similarly, based on the MAIA trial, FDA approved Dara-Rd for non-transplant-eligible NDMM (median PFS of about 5 years)[183]. In September 2019, the FDA also approved Dara in the first-line setting for transplant-eligible NDMM based on data from the CASSIOPEIA study (Dara-VTd)[184]. Dara was also investigated in combination with carfilzomib instead of bortezomib in the APOLLO study, which led to the approval in 2021 of Dara-pomalidomide/carfilzomib for RRMM[185].
Clinical trials investigating monoclonal antibodies in MM
| Drug - target molecule | Trial name | Study population | Outcomes | NCT |
| Daratumumab - CD38 | SIRIUS | RRMM | ORR 29%; mPFS 3.7 months | NCT01985126 |
| CASTOR | RRMM | ORR 83%; mPFS 16.7 months | NCT02136134 | |
| POLLUX | RRMM | ORR 92.9%; mPFS 44.5 months | NCT02076009 | |
| APOLLO | RRMM | mPFS 12.4 months | NCT03180736 | |
| ALCYONE | NTE NDMM | ORR 90.9%; mPFS 36.9 months | NCT02195479 | |
| MAIA | NTE NDMM | ORR 92.9% months; mPFS 5 years | NCT02252172 | |
| CASSIOPEIA | TE NDMM | ORR 92.6% CR 39% months; CR 54% | NCT02541383 | |
| Isatuximab - CD38 | ICARIA - MM | RRMM | ORR 60.4%; mPFS 11.5 months; mOS 24.6 months | NCT02990338 |
| IKEMA | RRMM | ORR 87% months | NCT03275285 | |
| IsKia | TE NDMM | NA | NCT04483739 | |
| Elotuzumab - SLAM7 | ELOQUENT-2 | RRMM | PFS 19.4 months, ORR 79% | NCT01239797 |
| ELOQUENT-3 | RRMM | PFS 10.2 months, ORR 53% | NCT02654132 |
Similar to daratumumab, isatuximab (isa) targets the CD38 receptor on a different epitope. The ICARIA-MM trial led to the approval of isa/pomalidomide/dexamethasone for RRMM (median PFS of 11.5 months)[186]. Similarly, the IKEMA study led to the approval of isa/carfilzomib/dexamethasone for RRMM (median PFS 35.7 months)[187]. Isatuximab is also being investigated upfront as part of the induction and consolidation regimens in the IsKia trial in transplant-eligible patients comparing Isa-KRd vs KRd (NCT04483739).
SLAM7 MAbs
Elotuzumab (E) is a monoclonal antibody that targets the signaling lymphocytic activation molecule family member 7 (SLAM7). SLAM7 is expressed on both MM and NK cells and exerts antitumor activity by activating NK cells directly and via CD16-mediated antibody-dependent cellular cytotoxicity (ADCC)[188]. ADCC occurs when the Fc receptors on NK cells bind to the Fc portion of elotuzumab and release toxic enzymes that kill the tumor cell[188]. Importantly, unlike daratumumab, although SLAM7 is expressed on NK cells, elotuzumab does not induce NK cell fratricide[188]. On the contrary, SLAM7 was detected at high levels on exhausted CD8+ T cells and elotuzumab was able to specifically eliminate these cells[189]. In 2015, following the ELOQUENT-2 trial, the FDA approved elotuzumab/lenalidomide/dexamethasone for RRMM (PFS 19.4 months, ORR 79%)[190]. In 2018, following the ELOQUENT-3 trial, the FDA approved elotuzumab/pomalidomide/dexamethasone for RRMM (PFS 10.2 months, ORR 53%)[191]. Recently, a phase 1 clinical trial has investigated the use of elotuzumab in conjunction with peripheral blood cells to boost NK cell function in the early post-transplant setting, showing no concerns for safety[192]. However, it was found that the suppressive MM microenvironment actively inhibits the function of immune cells, including NK cells, leading eventually to relapse[192].
CONCLUSION
Multiple myeloma is a clinically and molecularly heterogenous disorder, which is typically characterized by immunoparesis, involving both innate and adaptive compartment, contributing to disease recurrence and therapy resistance[193]. The evolution from MGUS to SMM to active MM is associated with editing of the immune context. Unfortunately, most of these changes remain to be fully elucidated. As MM development is strictly supported by the interaction with the BM microenvironment, treatment targeting this cross-talk is necessary to eradicate this disorder. Over the years, several approaches aimed at evoking the immune response against MM have been developed, although none of them so far appeared to be as revolutionary and successful in MM as some of them are in other tumor models. Although the reason for this lack of response remains elusive, the profound exhaustion of the immune system might play a pivotal role. In other words, in MM, there is no identifiable dominant immunosuppressive mechanism that can be appointed as a key target to restore immune competence. Instead, MM is characterized by a complicated and redundant network of molecular and metabolic axes that can compensate for each others’ functions when targeted by immunotherapeutic strategies. A potential therapeutic strategy would be using immunotherapy early when the immune system is still intact, and there is a low tumor burden disease in order to harness patients own immune system to target the cancer cells and prevent progression.
In recent years, incredible advancement has been made in understanding the biology of MM clones and the immune microenvironment. Studies thus far have shown that the biology of the immune microenvironment is heterogenous and different between distinct patients and plastic within the same patient upon therapy. Multiparametric flow cytometry employing several markers holds great promise in incorporating a comprehensive characterization of the immune environment into the design of clinical trials. For example, patients that show at diagnosis absence of an “exhausted” immune system may benefit from immunotherapy, such as CAR-T and BiTEs, upfront. Given the immense availability of more effective and tailored treatments in the near future, clinicians and scientists will be able to design more personalized therapeutic plans for myeloma patients with unique microenvironments.
DECLARATION
AcknowledgmentsGiada Bianchi and Maria Moscvin are thankful to the Demarest Lloyd Jr Foundation and the Appleby Cardiac Amyloidosis Fund for their support of the Amyloidosis Program.
Authors contributionsConceived the study; secured funding for the study: Bianchi G, Moscvin M
Wrote the first draft of the manuscript: Bianchi G, Moscvin M, Evans B
All authors revised and accepted the final version of the manuscript.
Availability of data and materialsNot applicable.
Financial support and sponsorshipThis work was supported in part by NIH/NCI K08CA245100 grant (Giada Bianchi), by the American Italian Cancer Foundation Research Fellowship (Maria Moscvin), and the Donald C. Brockman Memorial Research Grant from the Amyloidosis Foundation (Maria Moscvin).
Conflicts of interestGiada Bianchi: Advisory board participation (with personal payment): Pfizer, Karyopharm; Other authors declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationNot applicable.
Copyright© The Author(s) 2023.
REFERENCES
2. American Cancer Society. Cancer facts and figures; 2022. Available from: https://www.cancer.org/research/cancer-facts-statistics/all-cancer-facts-figures/cancer-facts-figures-2022.html [Last accessed on 16 May 2023].
3. Braunlin M, Belani R, Buchanan J, Wheeling T, Kim C. Trends in the multiple myeloma treatment landscape and survival: a U.S. analysis using 2011-2019 oncology clinic electronic health record data. Leuk Lymphoma 2021;62:377-86.
4. Bianchi G, Richardson PG, Anderson KC. Best treatment strategies in high-risk multiple myeloma: navigating a gray area. J Clin Oncol 2014;32:2125-32.
5. Lohr JG, Stojanov P, Carter SL, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell 2014;25:91-101.
6. Ledergor G, Weiner A, Zada M, et al. Single cell dissection of plasma cell heterogeneity in symptomatic and asymptomatic myeloma. Nat Med 2018;24:1867-76.
7. Merz M, Merz AMA, Wang J, et al. Deciphering spatial genomic heterogeneity at a single cell resolution in multiple myeloma. Nat Commun 2022;13:807.
8. Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 2009;113:5412-7.
9. Kyle RA, Durie BG, Rajkumar SV, et al. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010;24:1121-7.
10. Perrot A, Lauwers-Cances V, Tournay E, et al. Development and validation of a cytogenetic prognostic index predicting survival in multiple myeloma. J Clin Oncol 2019;37:1657-65.
11. Bolli N, Maura F, Minvielle S, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun 2018;9:3363.
12. Ho M, Patel A, Goh CY, Moscvin M, Zhang L, Bianchi G. Changing paradigms in diagnosis and treatment of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Leukemia 2020;34:3111-25.
13. Zavidij O, Haradhvala NJ, Mouhieddine TH, et al. Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer 2020;1:493-506.
14. Ramachandran IR, Martner A, Pisklakova A, et al. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol 2013;190:3815-23.
15. Görgün GT, Whitehill G, Anderson JL, et al. Tumor-promoting immune-suppressive myeloid-derived suppressor cells in the multiple myeloma microenvironment in humans. Blood 2013;121:2975-87.
16. Bronte V, Brandau S, Chen SH, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun 2016;7:12150.
17. Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood 2019;133:2159-67.
18. Rodriguez PC, Quiceno DG, Zabaleta J, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 2004;64:5839-49.
19. Darcy CJ, Minigo G, Piera KA, et al. Neutrophils with myeloid derived suppressor function deplete arginine and constrain T cell function in septic shock patients. Crit Care 2014;18:R163.
20. Bronte V, Serafini P, De Santo C, et al. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J Immunol 2003;170:270-8.
21. Mazzoni A, Bronte V, Visintin A, et al. Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 2002;168:689-95.
22. Malek E, de Lima M, Letterio JJ, et al. Myeloid-derived suppressor cells: the green light for myeloma immune escape. Blood Rev 2016;30:341-8.
23. Sawant A, Deshane J, Jules J, et al. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res 2013;73:672-82.
24. Zhuang J, Zhang J, Lwin ST, et al. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS One 2012;7:e48871.
25. Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016;128:384-94.
26. Casneuf T, Adams HC 3rd, van de Donk NWCJ, et al. Deep immune profiling of patients treated with lenalidomide and dexamethasone with or without daratumumab. Leukemia 2021;35:573-84.
27. Kuwahara-Ota S, Shimura Y, Steinebach C, et al. Lenalidomide and pomalidomide potently interfere with induction of myeloid-derived suppressor cells in multiple myeloma. Br J Haematol 2020;191:784-95.
28. Shaul ME, Fridlender ZG. Tumour-associated neutrophils in patients with cancer. Nat Rev Clin Oncol 2019;16:601-20.
29. Pillay J, Tak T, Kamp VM, Koenderman L. Immune suppression by neutrophils and granulocytic myeloid-derived suppressor cells: similarities and differences. Cell Mol Life Sci 2013;70:3813-27.
30. Romano A, Parrinello NL, Simeon V, et al. High-density neutrophils in MGUS and multiple myeloma are dysfunctional and immune-suppressive due to increased STAT3 downstream signaling. Sci Rep 2020;10:1983.
31. Romano A, Laura Parrinello N, Cerchione C, et al. The NLR and LMR ratio in newly diagnosed MM patients treated upfront with novel agents. Blood Cancer J 2017;7:649.
32. Lee GW, Park SW, Go SI, et al. The derived neutrophil-to-lymphocyte ratio is an independent prognostic factor in transplantation ineligible patients with multiple myeloma. Acta Haematol 2018;140:146-56.
33. Perez C, Botta C, Zabaleta A, et al. Immunogenomic identification and characterization of granulocytic myeloid-derived suppressor cells in multiple myeloma. Blood 2020;136:199-209.
34. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 2002;23:549-55.
35. Wu K, Lin K, Li X, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol 2020;11:1731.
36. Kim J, Denu RA, Dollar BA, et al. Macrophages and mesenchymal stromal cells support survival and proliferation of multiple myeloma cells. Br J Haematol 2012;158:336-46.
37. Zheng Y, Cai Z, Wang S, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood 2009;114:3625-8.
38. Chen X, Chen J, Zhang W, et al. Prognostic value of diametrically polarized tumor-associated macrophages in multiple myeloma. Oncotarget 2017;8:112685-96.
39. Zheng Y, Yang J, Qian J, et al. PSGL-1/selectin and ICAM-1/CD18 interactions are involved in macrophage-induced drug resistance in myeloma. Leukemia 2013;27:702-10.
40. Hope C, Ollar SJ, Heninger E, et al. TPL2 kinase regulates the inflammatory milieu of the myeloma niche. Blood 2014;123:3305-15.
41. Storti P, Bolzoni M, Donofrio G, et al. Hypoxia-inducible factor (HIF)-1α suppression in myeloma cells blocks tumoral growth in vivo inhibiting angiogenesis and bone destruction. Leukemia 2013;27:1697-706.
42. Rao L, Giannico D, Leone P, et al. HB-EGF-EGFR signaling in bone marrow endothelial cells mediates angiogenesis associated with multiple myeloma. Cancers 2020;12:173.
43. Filippi I, Saltarella I, Aldinucci C, et al. Different adaptive responses to hypoxia in normal and multiple myeloma endothelial cells. Cell Physiol Biochem 2018;46:203-12.
44. Scavelli C, Nico B, Cirulli T, et al. Vasculogenic mimicry by bone marrow macrophages in patients with multiple myeloma. Oncogene 2008;27:663-74.
45. Podar K, Anderson KC. The pathophysiologic role of VEGF in hematologic malignancies: therapeutic implications. Blood 2005;105:1383-95.
46. Somlo G, Lashkari A, Bellamy W, et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: a California cancer consortium trial. Br J Haematol 2011;154:533-5.
47. White D, Kassim A, Bhaskar B, Yi J, Wamstad K, Paton VE. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer 2013;119:339-47.
48. Beyar-Katz O, Magidey K, Reiner-Benaim A, et al. Proinflammatory macrophages promote multiple myeloma resistance to bortezomib therapy. Mol Cancer Res 2019;17:2331-40.
49. Mougiakakos D, Bach C, Böttcher M, et al. The IKZF1-IRF4/IRF5 axis controls polarization of myeloma-associated macrophages. Cancer Immunol Res 2021;9:265-78.
50. Krönke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 2014;343:301-5.
51. Wang Q, Lu Y, Li R, et al. Therapeutic effects of CSF1R-blocking antibodies in multiple myeloma. Leukemia 2018;32:176-83.
52. Brown JM, Recht L, Strober S. The promise of targeting macrophages in cancer therapy. Clin Cancer Res 2017;23:3241-50.
53. Gutiérrez-González A, Martínez-Moreno M, Samaniego R, et al. Evaluation of the potential therapeutic benefits of macrophage reprogramming in multiple myeloma. Blood 2016;128:2241-52.
54. Kim D, Wang J, Willingham SB, Martin R, Wernig G, Weissman IL. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia 2012;26:2538-45.
55. Chen H, Li M, Sanchez E, et al. JAK1/2 pathway inhibition suppresses M2 polarization and overcomes resistance of myeloma to lenalidomide by reducing TRIB1, MUC1, CD44, CXCL12, and CXCR4 expression. Br J Haematol 2020;188:283-94.
56. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med 1973;137:1142-62.
57. Rossi M, Young JW. Human dendritic cells: potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J Immunol 2005;175:1373-81.
58. Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol 2012;12:557-69.
59. Hideshima T, Mitsiades C, Tonon G, Richardson PG, Anderson KC. Understanding multiple myeloma pathogenesis in the bone marrow to identify new therapeutic targets. Nat Rev Cancer 2007;7:585-98.
60. Ratta M, Fagnoni F, Curti A, et al. Dendritic cells are functionally defective in multiple myeloma: the role of interleukin-6. Blood 2002;100:230-7.
61. Yang DH, Park JS, Jin CJ, et al. The dysfunction and abnormal signaling pathway of dendritic cells loaded by tumor antigen can be overcome by neutralizing VEGF in multiple myeloma. Leuk Res 2009;33:665-70.
62. Chauhan D, Singh AV, Brahmandam M, et al. Functional interaction of plasmacytoid dendritic cells with multiple myeloma cells: a therapeutic target. Cancer Cell 2009;16:309-23.
63. Moser-Katz T, Joseph NS, Dhodapkar MV, Lee KP, Boise LH. Game of bones: how myeloma manipulates its microenvironment. Front Oncol 2020;10:625199.
64. Leone P, Berardi S, Frassanito MA, et al. Dendritic cells accumulate in the bone marrow of myeloma patients where they protect tumor plasma cells from CD8+ T-cell killing. Blood 2015;126:1443-51.
65. Ray A, Das DS, Song Y, et al. Targeting PD1-PDL1 immune checkpoint in plasmacytoid dendritic cell interactions with T cells, natural killer cells and multiple myeloma cells. Leukemia 2015;29:1441-4.
66. Talluri S, Samur MK, Buon L, et al. Dysregulated APOBEC3G causes DNA damage and promotes genomic instability in multiple myeloma. Blood Cancer J 2021;11:166.
67. Koduru S, Wong E, Strowig T, et al. Dendritic cell-mediated activation-induced cytidine deaminase (AID)-dependent induction of genomic instability in human myeloma. Blood 2012;119:2302-9.
68. Prabhala RH, Fulciniti M, Pelluru D, et al. Targeting IL-17A in multiple myeloma: a potential novel therapeutic approach in myeloma. Leukemia 2016;30:379-89.
69. Murray ME, Gavile CM, Nair JR, et al. CD28-mediated pro-survival signaling induces chemotherapeutic resistance in multiple myeloma. Blood 2014;123:3770-9.
70. Stocker N, Gaugler B, Ricard L, et al. Daratumumab prevents programmed death ligand-1 expression on antigen-presenting cells in de novo multiple myeloma. Cancer Med 2020;9:2077-84.
71. Bailur JK, McCachren SS, Doxie DB, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight 2019;5:127807.
72. Feyler S, von Lilienfeld-Toal M, Jarmin S, et al. CD4+CD25+FoxP3+regulatory T cells are increased whilst CD3+CD4−CD8−αβTCR+Double Negative T cells are decreased in the peripheral blood of patients with multiple myeloma which correlates with disease burden. Br J Haematol 2009;144:686-95.
73. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003;4:330-6.
74. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol 2008;8:523-32.
75. Giannopoulos K, Kaminska W, Hus I, Dmoszynska A. The frequency of T regulatory cells modulates the survival of multiple myeloma patients: detailed characterisation of immune status in multiple myeloma. Br J Cancer 2012;106:546-52.
76. Dahlhoff J, Manz H, Steinfatt T, et al. Transient regulatory T-cell targeting triggers immune control of multiple myeloma and prevents disease progression. Leukemia 2022;36:790-800.
77. Feyler S, Scott GB, Parrish C, et al. Tumour cell generation of inducible regulatory T-cells in multiple myeloma is contact-dependent and antigen-presenting cell-independent. PLoS One 2012;7:e35981.
78. Tamura H, Ishibashi M, Yamashita T, et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013;27:464-72.
79. Yousef S, Marvin J, Steinbach M, et al. Immunomodulatory molecule PD-L1 is expressed on malignant plasma cells and myeloma-propagating pre-plasma cells in the bone marrow of multiple myeloma patients. Blood Cancer J 2015;5:e285.
80. Liu J, Hamrouni A, Wolowiec D, et al. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007;110:296-304.
81. Tai YT, Lin L, Xing L, et al. APRIL signaling via TACI mediates immunosuppression by T regulatory cells in multiple myeloma: therapeutic implications. Leukemia 2019;33:426-38.
82. Tai YT, Acharya C, An G, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016;127:3225-36.
83. Scott GB, Carter C, Parrish C, Wood PM, Cook G. Downregulation of myeloma-induced ICOS-L and regulatory T cell generation by lenalidomide and dexamethasone therapy. Cell Immunol 2015;297:1-9.
84. Hadjiaggelidou C, Mandala E, Terpos E, et al. Evaluation of regulatory T cells (Tregs) alterations in patients with multiple myeloma treated with bortezomib or lenalidomide plus dexamethasone: correlations with treatment outcome. Ann Hematol 2019;98:1457-66.
85. Dosani T, Carlsten M, Maric I, Landgren O. The cellular immune system in myelomagenesis: NK cells and T cells in the development of MM and their uses in immunotherapies. Blood Cancer J 2015;5:e321.
86. Favaloro J, Brown R, Aklilu E, et al. Myeloma skews regulatory T and pro-inflammatory T helper 17 cell balance in favor of a suppressive state. Leuk Lymphoma 2014;55:1090-8.
87. Shen CJ, Yuan ZH, Liu YX, Hu GY. Increased numbers of T helper 17 cells and the correlation with clinicopathological characteristics in multiple myeloma. J Int Med Res 2012;40:556-64.
88. Noonan K, Marchionni L, Anderson J, Pardoll D, Roodman GD, Borrello I. A novel role of IL-17-producing lymphocytes in mediating lytic bone disease in multiple myeloma. Blood 2010;116:3554-63.
89. Rossi M, Altomare E, Botta C, et al. miR-21 antagonism abrogates Th17 tumor promoting functions in multiple myeloma. Leukemia 2021;35:823-34.
90. Suen H, Brown R, Yang S, et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia 2016;30:1716-24.
91. Racanelli V, Leone P, Frassanito MA, et al. Alterations in the antigen processing-presenting machinery of transformed plasma cells are associated with reduced recognition by CD8+ T cells and characterize the progression of MGUS to multiple myeloma. Blood 2010;115:1185-93.
92. Zelle-Rieser C, Thangavadivel S, Biedermann R, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol 2016;9:116.
93. Dhodapkar MV, Krasovsky J, Olson K. T cells from the tumor microenvironment of patients with progressive myeloma can generate strong, tumor-specific cytolytic responses to autologous, tumor-loaded dendritic cells. Proc Natl Acad Sci USA 2002;99:13009-13.
94. Wen YJ, Min R, Tricot G, Barlogie B, Yi Q. Tumor lysate-specific cytotoxic T lymphocytes in multiple myeloma: promising effector cells for immunotherapy. Blood 2002;99:3280-5.
95. Guillerey C, Nakamura K, Vuckovic S, Hill GR, Smyth MJ. Immune responses in multiple myeloma: role of the natural immune surveillance and potential of immunotherapies. Cell Mol Life Sci 2016;73:1569-89.
96. Eltzschig HK, Sitkovsky MV, Robson SC. Purinergic signaling during inflammation. N Engl J Med 2012;367:2322-33.
97. Deaglio S, Dwyer KM, Gao W, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 2007;204:1257-65.
98. Horenstein AL, Quarona V, Toscani D, et al. Adenosine generated in the bone marrow niche through a CD38-Mediated pathway correlates with progression of human myeloma. Mol Med 2016;22:694-704.
99. Yang R, Elsaadi S, Misund K, et al. Conversion of ATP to adenosine by CD39 and CD73 in multiple myeloma can be successfully targeted together with adenosine receptor A2A blockade. J Immunother Cancer 2020;8:e000610.
100. Bonello F, D'Agostino M, Moscvin M, Cerrato C, Boccadoro M, Gay F. CD38 as an immunotherapeutic target in multiple myeloma. Expert Opin Biol Ther 2018;18:1209-21.
101. Mateos MV, Blacklock H, Schjesvold F, et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): a randomised, open-label, phase 3 trial. Lancet Haematol 2019;6:e459-69.
102. Usmani SZ, Schjesvold F, Oriol A, et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): a randomised, open-label, phase 3 trial. Lancet Haematol 2019;6:e448-58.
103. Guillerey C, Harjunpää H, Carrié N, et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood 2018;132:1689-94.
104. Lozano E, Mena MP, Díaz T, et al. Nectin-2 expression on malignant plasma cells is associated with better response to TIGIT blockade in multiple myeloma. Clin Cancer Res 2020;26:4688-98.
105. Minnie SA, Kuns RD, Gartlan KH, et al. Myeloma escape after stem cell transplantation is a consequence of T-cell exhaustion and is prevented by TIGIT blockade. Blood 2018;132:1675-88.
106. García-Sanz R, González M, Orfão A, et al. Analysis of natural killer-associated antigens in peripheral blood and bone marrow of multiple myeloma patients and prognostic implications. Br J Haematol 1996;93:81-8.
107. Pessoa de Magalhães RJ, Vidriales MB, Paiva B, et al. Analysis of the immune system of multiple myeloma patients achieving long-term disease control by multidimensional flow cytometry. Haematologica 2013;98:79-86.
108. Schütt P, Brandhorst D, Stellberg W, et al. Immune parameters in multiple myeloma patients: influence of treatment and correlation with opportunistic infections. Leuk Lymphoma 2006;47:1570-82.
109. Fauriat C, Mallet F, Olive D, Costello RT. Impaired activating receptor expression pattern in natural killer cells from patients with multiple myeloma. Leukemia 2006;20:732-3.
110. Jurisic V, Srdic T, Konjevic G, Markovic O, Colovic M. Clinical stage-depending decrease of NK cell activity in multiple myeloma patients. Med Oncol 2007;24:312-7.
111. Botta C, Mendicino F, Martino EA, et al. Mechanisms of immune evasion in multiple myeloma: open questions and therapeutic opportunities. Cancers 2021;13:3213.
112. Bedel R, Thiery-Vuillemin A, Grandclement C, et al. Novel role for STAT3 in transcriptional regulation of NK immune cell targeting receptor MICA on cancer cells. Cancer Res 2011;71:1615-26.
113. Jinushi M, Vanneman M, Munshi NC, et al. MHC class I chain-related protein A antibodies and shedding are associated with the progression of multiple myeloma. Proc Natl Acad Sci USA 2008;105:1285-90.
114. El-Sherbiny YM, Meade JL, Holmes TD, et al. The requirement for DNAM-1, NKG2D, and NKp46 in the natural killer cell-mediated killing of myeloma cells. Cancer Res 2007;67:8444-9.
115. Fisher DT, Appenheimer MM, Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol 2014;26:38-47.
116. Cifaldi L, Prencipe G, Caiello I, et al. Inhibition of natural killer cell cytotoxicity by interleukin-6: implications for the pathogenesis of macrophage activation syndrome. Arthritis Rheumatol 2015;67:3037-46.
117. Beldi-Ferchiou A, Lambert M, Dogniaux S, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016;7:72961-77.
118. Lesokhin AM, Ansell SM, Armand P, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol 2016;34:2698-704.
119. Shah N, Li L, McCarty J, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol 2017;177:457-66.
120. Shah N, Mehta R, Li L, et al. Phase II study of
121. Romee R, Schneider SE, Leong JW, et al. Cytokine activation induces human memory-like NK cells. Blood 2012;120:4751-60.
122. Ho M, Goh CY, Patel A, et al. Role of the bone marrow milieu in multiple myeloma progression and therapeutic resistance. Clin Lymphoma Myeloma Leuk 2020;20:e752-68.
123. Bianchi G, Munshi NC. Pathogenesis beyond the cancer clone(s) in multiple myeloma. Blood 2015;125:3049-58.
124. de Jong MME, Kellermayer Z, Papazian N, et al. The multiple myeloma microenvironment is defined by an inflammatory stromal cell landscape. Nat Immunol 2021;22:769-80.
125. Abe M, Hiura K, Wilde J, et al. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood 2004;104:2484-91.
126. An G, Acharya C, Feng X, et al. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: therapeutic implication. Blood 2016;128:1590-603.
127. Giuliani N, Colla S, Sala R, et al. Human myeloma cells stimulate the receptor activator of nuclear factor-kappa B ligand (RANKL) in T lymphocytes: a potential role in multiple myeloma bone disease. Blood 2002;100:4615-21.
128. Giuliani N, Morandi F, Tagliaferri S, et al. Interleukin-3 (IL-3) is overexpressed by T lymphocytes in multiple myeloma patients. Blood 2006;107:841-2.
129. Chen T, Moscvin M, Bianchi G. Exosomes in the pathogenesis and treatment of multiple myeloma in the context of the bone marrow microenvironment. Front Oncol 2020;10:608815.
130. Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009;9:581-93.
131. Niel G, D'Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 2018;19:213-28.
132. Colombo M, Raposo G, Théry C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 2014;30:255-89.
133. Peinado H, Lavotshkin S, Lyden D. The secreted factors responsible for pre-metastatic niche formation: old sayings and new thoughts. Semin Cancer Biol 2011;21:139-46.
134. Ratajczak J, Wysoczynski M, Hayek F, Janowska-Wieczorek A, Ratajczak MZ. Membrane-derived microvesicles: important and underappreciated mediators of cell-to-cell communication. Leukemia 2006;20:1487-95.
135. Lemaire M, Deleu S, De Bruyne E, Van Valckenborgh E, Menu E, Vanderkerken K. The microenvironment and molecular biology of the multiple myeloma tumor. Adv Cancer Res 2011;110.
136. Boyiadzis M, Whiteside TL. The emerging roles of tumor-derived exosomes in hematological malignancies. Leukemia 2017;31:1259-68.
137. Moloudizargari M, Abdollahi M, Asghari MH, Zimta AA, Neagoe IB, Nabavi SM. The emerging role of exosomes in multiple myeloma. Blood Rev 2019;38:100595.
138. Li M, Xia B, Wang Y, You MJ, Zhang Y. Potential therapeutic roles of exosomes in multiple myeloma: a systematic review. J Cancer 2019;10:6154-60.
139. Roccaro AM, Sacco A, Maiso P, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest 2013;123:1542-55.
140. Xu H, Han H, Song S, et al. Exosome-transmitted PSMA3 and PSMA3-AS1 promote proteasome inhibitor resistance in multiple myeloma. Clin Cancer Res 2019;25:1923-35.
141. Ho M, Chen T, Liu J, et al. Targeting histone deacetylase 3 (HDAC3) in the bone marrow microenvironment inhibits multiple myeloma proliferation by modulating exosomes and IL-6 trans-signaling. Leukemia 2020;34:196-209.
142. Raimondi L, De Luca A, Giavaresi G, et al. Non-coding RNAs in multiple myeloma bone disease pathophysiology. Noncoding RNA 2020;6:37.
143. Morelli E, Gullà A, Rocca R, et al. The non-coding RNA landscape of plasma cell dyscrasias. Cancers 2020;12:320.
144. Li B, Xu H, Han H, et al. Exosome-mediated transfer of lncRUNX2-AS1 from multiple myeloma cells to MSCs contributes to osteogenesis. Oncogene 2018;37:5508-19.
145. Raimondo S, Saieva L, Vicario E, et al. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J Hematol Oncol 2019;12:2.
146. Raimondo S, Urzì O, Conigliaro A, et al. Extracellular vesicle microRNAs contribute to the osteogenic inhibition of mesenchymal stem cells in multiple myeloma. Cancers 2020;12:449.
147. Liu Z, Liu H, Li Y, et al. Multiple myeloma-derived exosomes inhibit osteoblastic differentiation and improve IL-6 secretion of BMSCs from multiple myeloma. J Investig Med 2020;68:45-51.
148. Liu R, Gao Q, Foltz SM, et al. Co-evolution of tumor and immune cells during progression of multiple myeloma. Nat Commun 2021;12:2559.
149. Calcinotto A, Ponzoni M, Ria R, et al. Modifications of the mouse bone marrow microenvironment favor angiogenesis and correlate with disease progression from asymptomatic to symptomatic multiple myeloma. Oncoimmunology 2015;4:e1008850.
150. Hofmann JN, Landgren O, Landy R, et al. A prospective study of circulating chemokines and angiogenesis markers and risk of multiple myeloma and its precursor. JNCI Cancer Spectr 2020;4:pkz104.
151. Knight A, Rihova L, Kralova R, et al. Plasmacytoid dendritic cells in patients with MGUS and multiple myeloma. J Clin Med 2021;10:3717.
152. Dhodapkar MV, Krasovsky J, Osman K, Geller MD. Vigorous premalignancy-specific effector T cell response in the bone marrow of patients with monoclonal gammopathy. J Exp Med 2003;198:1753-7.
153. Calcinotto A, Brevi A, Chesi M, et al. Microbiota-driven interleukin-17-producing cells and eosinophils synergize to accelerate multiple myeloma progression. Nat Commun 2018;9:4832.
154. Bernal M, Garrido P, Jiménez P, et al. Changes in activatory and inhibitory natural killer (NK) receptors may induce progression to multiple myeloma: implications for tumor evasion of T and NK cells. Hum Immunol 2009;70:854-7.
155. Bernardini G, Sciumè G, Santoni A. Differential chemotactic receptor requirements for NK cell subset trafficking into bone marrow. Front Immunol 2013;4:12.
156. Ponzetta A, Benigni G, Antonangeli F, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Cancer Res 2015;75:4766-77.
157. Bobin A, Leleu X. Recent advances in the treatment of multiple myeloma: a brief review. Fac Rev 2022;11:28.
158. Munshi NC, Anderson LD Jr, Shah N, et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 2021;384:705-16.
159. Anderson LD Jr. Idecabtagene vicleucel (ide-cel) CAR T-cell therapy for relapsed and refractory multiple myeloma. Future Oncol 2022;18:277-89.
160. Rodriguez-Otero P, Ailawadhi S, Arnulf B, et al. Ide-cel or standard regimens in relapsed and refractory multiple myeloma. N Engl J Med 2023;388:1002-14.
161. Martin T, Usmani SZ, Berdeja JG, et al. Ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol 2023;41:1265-74.
162. Van Oekelen O, Nath K, Mouhieddine TH, et al. Interventions and outcomes of patients with multiple myeloma receiving salvage therapy after BCMA-directed CAR T therapy. Blood 2023;141:756-65.
163. Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol 2020;17:147-67.
164. Teoh PJ, Chng WJ. CAR T-cell therapy in multiple myeloma: more room for improvement. Blood Cancer J 2021;11:84.
165. Ho M, Xiao A, Yi D, Zanwar S, Bianchi G. Treating multiple myeloma in the context of the bone marrow microenvironment. Curr Oncol 2022;29:8975-9005.
166. Tian Z, Liu M, Zhang Y, Wang X. Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. J Hematol Oncol 2021;14:75.
168. Moreau P, Girgis S, Goldberg JD. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med 2022;387:1721-3.
169. Bahlis NJ, Raje NS, Costello C, et al. Efficacy and safety of elranatamab (PF-06863135), a B-cell maturation antigen (BCMA)-CD3 bispecific antibody, in patients with relapsed or refractory multiple myeloma (MM). J Clin Oncol 2021;39:8006-8006.
170. Jakubowiak AJ, Bahlis NJ, Raje NS, et al. Elranatamab, a BCMA-targeted T-cell redirecting immunotherapy, for patients with relapsed or refractory multiple myeloma: updated results from MagnetisMM-1. J Clin Oncol 2022;40:8014-8014.
171. Verkleij CPM, Broekmans MEC, van Duin M, et al. Preclinical activity and determinants of response of the GPRC5DxCD3 bispecific antibody talquetamab in multiple myeloma. Blood Adv 2021;5:2196-215.
172. Pillarisetti K, Edavettal S, Mendonça M, et al. A T-cell-redirecting bispecific G-protein-coupled receptor class 5 member D x CD3 antibody to treat multiple myeloma. Blood 2020;135:1232-43.
173. Doucey M, Pouleau B, Estoppey C, et al. ISB 1342: a first-in-class CD38 T cell engager for the treatment of relapsed refractory multiple myeloma. J Clin Oncol 2021;39:8044-8044.
174. Lonial S, Lee HC, Badros A, et al. Longer term outcomes with single-agent belantamab mafodotin in patients with relapsed or refractory multiple myeloma: 13-month follow-up from the pivotal DREAMM-2 study. Cancer 2021;127:4198-212.
175. Ailawadhi S, Kelly KR, Vescio RA, et al. A phase I study to assess the safety and pharmacokinetics of single-agent lorvotuzumab mertansine (IMGN901) in patients with relapsed and/or refractory CD-56-positive multiple myeloma. Clin Lymphoma Myeloma Leuk 2019;19:29-34.
176. Kelly KR, Ailawadhi S, Siegel DS, et al. Indatuximab ravtansine plus dexamethasone with lenalidomide or pomalidomide in relapsed or refractory multiple myeloma: a multicentre, phase 1/2a study. Lancet Haematol 2021;8:e794-807.
177. Fionda C, Abruzzese MP, Zingoni A, et al. The IMiDs targets IKZF-1/3 and IRF4 as novel negative regulators of NK cell-activating ligands expression in multiple myeloma. Oncotarget 2015;6:23609-30.
178. Feng X, Zhang L, Acharya C, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res 2017;23:4290-300.
179. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet 2016;387:1551-60.
180. Mateos MV, Sonneveld P, Hungria V, et al. Daratumumab, bortezomib, and dexamethasone versus bortezomib and dexamethasone in patients with previously treated multiple myeloma: three-year follow-up of CASTOR. Clin Lymphoma Myeloma Leuk 2020;20:509-18.
181. Bahlis NJ, Dimopoulos MA, White DJ, et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020;34:1875-84.
182. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N Engl J Med 2018;378:518-28.
183. Facon T, Kumar S, Plesner T, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med 2019;380:2104-15.
184. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet 2019;394:29-38.
185. Dimopoulos MA, Terpos E, Boccadoro M, et al. Daratumumab plus pomalidomide and dexamethasone versus pomalidomide and dexamethasone alone in previously treated multiple myeloma (APOLLO): an open-label, randomised, phase 3 trial. Lancet Oncol 2021;22:801-12.
186. Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet 2019;394:2096-107.
187. Moreau P, Dimopoulos MA, Mikhael J, et al. Isatuximab, carfilzomib, and dexamethasone in relapsed multiple myeloma (IKEMA): a multicentre, open-label, randomised phase 3 trial. Lancet 2021;397:2361-71.
188. Campbell KS, Cohen AD, Pazina T. Mechanisms of NK cell activation and clinical activity of the therapeutic SLAMF7 antibody, elotuzumab in multiple myeloma. Front Immunol 2018;9:2551.
189. Awwad MHS, Mahmoud A, Bruns H, et al. Selective elimination of immunosuppressive T cells in patients with multiple myeloma. Leukemia 2021;35:2602-15.
190. Lonial S, Dimopoulos M, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015;373:621-31.
191. Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N Engl J Med 2018;379:1811-22.
192. Osman K, Chari A, Parekh S, et al. Phase 1 study of elotuzumab in combination with autologous stem cell transplantation and lenalidomide maintenance for multiple myeloma. Biol Blood Marrow Transplant 2018;24:S49-50.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Moscvin M, Evans B, Bianchi G. Dissecting molecular mechanisms of immune microenvironment dysfunction in multiple myeloma and precursor conditions. J Cancer Metastasis Treat 2023;9:17. http://dx.doi.org/10.20517/2394-4722.2022.110
AMA Style
Moscvin M, Evans B, Bianchi G. Dissecting molecular mechanisms of immune microenvironment dysfunction in multiple myeloma and precursor conditions. Journal of Cancer Metastasis and Treatment. 2023; 9: 17. http://dx.doi.org/10.20517/2394-4722.2022.110
Chicago/Turabian Style
Moscvin, Maria, Benjamin Evans, Giada Bianchi. 2023. "Dissecting molecular mechanisms of immune microenvironment dysfunction in multiple myeloma and precursor conditions" Journal of Cancer Metastasis and Treatment. 9: 17. http://dx.doi.org/10.20517/2394-4722.2022.110
ACS Style
Moscvin, M.; Evans B.; Bianchi G. Dissecting molecular mechanisms of immune microenvironment dysfunction in multiple myeloma and precursor conditions. J. Cancer. Metastasis. Treat. 2023, 9, 17. http://dx.doi.org/10.20517/2394-4722.2022.110
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 5 clicks
Cite This Article 5 clicks

 Like This Article 6
likes
Like This Article 6
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.