
Environmental aging of the skin: new insights

Abstract
The appearance of aging is determined primarily by extrinsic factors through exposure to environmental sunlight and airborne pollution. That solar ultraviolet B (λ = 290-320 nm) directly causes photoaging (with wrinkles, dryness, and mottled pigmentation) and skin cancer has been recognized for decades; the contribution by ultraviolet A (λ = 320-400 nm) was only more recently understood. New research further implicates visible light (λ = 400-700 nm) as well as the heat rays of infrared radiation (λ > 800 nm). Particularly in urban environments, airborne pollutants such as ozone (O3), polycyclic aromatic hydrocarbons, particulate matter (PM) in smog, and tobacco smoke contribute to photoaging and skin cancer. Furthermore, exposure simultaneously to both solar ultraviolet (UV) and these pollutants results in even greater synergistic damage. The volatile pollutants generate reactive oxygen species which oxidize surface lipids leading to deeper damaging inflammatory reactions. PM carries high concentrations of environmental organic compounds and trace metals. These pollutant-laden particles deliver toxins to the skin transcutaneously through hair follicles and through the blood after respiratory inhalation. The predominant natural mechanism of clearing these xenobiotic chemicals is through the ligand-activated transcription factor the arylhydrocarbon receptor (AHR) found on all skin cells. AHR activity regulates keratinocyte differentiation and proliferation, maintenance of epidermal barrier function, melanogenesis, and immunity. With chronic activation by UV exposure and pollutants, AHR signaling contributes to both extrinsic aging and carcinogenesis.
Keywords
Introduction
Today we live far longer than those in generations before us. The life expectancy of an individual born after 1990 is about 80 years, a substantial increase over the life expectancy of 65 years in those born in 1945. According to the 2018 United States Census Bureau Projections, of the approximately 36% of our present population now over 65 years old, at least a quarter will live to be over 95 years old. Thus it is imperative that our medical research goals emphasize not only lifespan, but also healthspan - maintaining our energy, mind, body, and happiness for all of these extra years.
Tantamount to healthspan is protecting our skin, the largest organ of our body and the only organ we see - our calling card to the world and shell of interface with external insults. We must be vigilant in protecting our skin from the external onslaught of environmental damage from solar ultraviolet (UV), visible light (VL), and infrared (IR) radiation and airborne pollutants. Science and technology have given us more leisure time so that we can travel to sunny climates and high altitudes where we stay outdoors for many hours swimming, sailing, or skiing, exposing ourselves not only to direct sunlight, but also virtually doubling our UV exposure from the indirect reflection of water, sand, and snow. Also we increasingly live in cities where we are surrounded by the ubiquitous pollutants of traffic and industry outdoors, and cigarette smoke, cleaning agents, and heating and cooking fuel indoors. All of these pollutants - including ozone (O3), polycyclic aromatic hydrocarbons (PAHs), volatile organic compounds (VOCs), and particulate matter (PM) - cause direct oxidative damage that leads to inflammatory responses, resulting in unattractive premature aging of the skin and deforming, dangerous skin cancer. Furthermore, recent research has proven that UV exposure not only directly harms the skin, but synergistically enhances the damage rendered by airborne pollutants[1].
Recent research has elucidated the precise cellular and molecular mechanisms of the cutaneous damage and of the innate destructive as well as protective responses. A new understanding of how the arylhydrocarbon receptor (AHR) processes xenobiotic insults from UV exposure as well as from airborne pollutants is explained. New evidence demonstrates that topical antioxidants (especially vitamin C, vitamin E, and L-selenomethionine) can indeed not only prevent but also partially reverse extrinsic aging of the skin, though stringent criteria in formulation are required to achieve and maintain optimal delivery, activity, and stability.
Solar photoaging
The first protective response of skin to solar UV radiation is production of melanin. After exposure to UV radiation, there is an immediate increase in tyrosinase activity accompanied by an increase in the size of melanosomes. Subsequent exposure results in an increase in the number of active melanocytes with increased transfer of Stage IV melanosomes to keratinocytes. The density of melanosomes in chronically sun-exposed skin is up to double that of non-sun exposed skin[2]. Within minutes after exposure to ultraviolet A (UVA) radiation, there is immediate pigmentary darkening (IPD) - probably due to oxidation of pre-existing melanin and melanin precursors. Because this fades within 20-30 min, it provides no protection. Protective delayed tanning is seen 24 h to 72 h after exposure to UVA + ultraviolet B (UVB) by the mechanisms described above[3].
All tanning is actually photodamage - extrinsic aging of the skin. In children, the delayed tanning fades within several weeks after terminating summer exposure, though some freckles may persist. However, individuals with no visible hyperpigmentation show the subclinical photodamage of hyperpigmentation when examined with a medical UVA Wood’s light (λ = 367 nm), as seen in the 45-year-old woman shown in Figure 1[3]. With chronic UV exposure, this hyperpigmentation becomes the clearly visible, with unattractive solar lentigines seen commonly in older individuals, especially outdoor sports enthusiasts or those with outdoor professions.
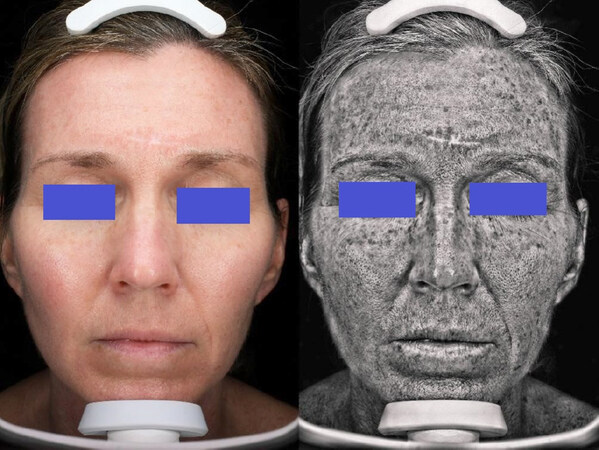
Figure 1. Subclinical photodamage in a 45-year-old female. No photodamage of hyperpigmentation can be seen unless examined with a Wood’s light (λ = 367 nm), revealing hyperpigmentation from previous sun exposure. VISIA ultraviolet photograph. (Printed with permission by Canfield Imaging Systems, Parsippany, NJ 07054.)
Individuals with light, sun-sensitive skin (Fitzpatrick phototypes I and II) are at high risk for UV-induced photoaging and skin cancer. The pheomelanin synthesized in response to UV actually produces reactive oxygen species (ROS), leading to increased photoaging and skin cancers, especially in red-heads. Also, damaging DNA photoproducts are produced by chemo-excitation of melanin derivatives, even long after UV exposure[4]. In addition, most red-haired individuals have dysfunction of their melanocortin-1 receptor (MC1R) which hampers the possibility of developing a tan.
Studies with confocal microscopy have demonstrated that individuals of Fitzpatrick skin types I and II not only have lower melanin but also significantly fewer dermal papillae than those with higher Fitzpatrick skin types and darker skin[5]. With age, there is an even greater disparity between light and dark skin in density of dermal papillae, with increasingly fewer in light-skinned individuals as they age. This is a less-recognized manifestation of photoaging, probably contributing to the crepe-like texture of extrinsically aged skin in light-skinned phototypes.
UV radiation
Only about 5% of solar terrestrial radiation is UV radiation. Of the UV component, 95% is UVA and 5% is UVB. The primary chromophore of UVB (λ = 290-320 nm) radiation is DNA which suffers specific “UVB fingerprint mutations” resulting in intrastrand pyrimidine dimers[6]. Further mutations are incited by the generation of ROS. With cumulative UVB damage and natural aging, there is a decreased ability for nucleotide excision repair so that mutations persist, directly leading to pre-cancerous actinic keratoses and skin cancers[6] - basal cell carcinomas (BCCs), squamous cell carcinomas (SCCs), and malignant melanomas (MMs) - of which SSCs and MMs are potentially lethal.
Ninety-five percent of our daily UV exposure is UVA (λ = 320-450 nm) which, unlike UVB, is not filtered by glass and does not vary seasonally in intensity. Like UVB, UVA exposure results in “signature mutations” with T → G transversions and formation of specific pyrimidine dimers[6]. UVA generates ROS with resultant DNA mutations, in particular oxidation of guanosine to 8-hydroxy-deoxyguanosine (8-OHdG). UVA penetrates the skin more deeply than does UVB: UVA reaches the epidermal stem-cell rich basal layer and the dermis to inactivate the tumor suppressor p53 gene and to decrease immune functions so that early skin cancers are not recognized. Only extremely high exposure to UVA might directly initiate skin cancer, while the lower doses of UVA to which we are exposed do inhibit the normal immune response so that UVB-initiated skin carcinomas proliferate unchecked. Furthermore, it is UVA that activates dermal matrix metalloproteinases (MMP) to degrade collagen and elastic tissue, resulting in the wrinkled, crepe-like, and saggy quality of photo-damaged skin.
High-energy visible blue light
Of the solar radiation that reaches the earth’s surface, 39% is visible light (VL, with λ = 400-700 nm) with the high-energy VL in the low wavelength range (blue light). We are exposed to the high-energy VL not only from sunlight but also from computer screens and smart phones! Early observations by Kollias et al.[7] showed that VL does induce skin pigmentation that can last for 10 weeks. Further research demonstrated by exposure to a xenon-mercury lamp, that VL (up to 470 nm) induces IPD, as does UVA-I[8]. The peak IPD response is at wavelengths λ = 300-500 nm, including all UVA-II (λ = 320-340 nm), UVA-I (λ = 340-400 nm), and visible blue light (λ = 400-500 nm)[9]. This tanning (with erythema) was only seen in dark-skinned individuals of Fitzpatrick skin type IV-VI, probably because of the large amount of melanin in their skin; light-skin type I individuals showed no tanning after VL exposure[10]. The corresponding coinciding erythema may be because as melanin absorbs VL, thereby generating heat to cause vasodilation. More exposure to VL leads to darker and more sustained pigmentation as seen also in UV-induced post-inflammatory hyperpigmentation.
Exposure of human skin equivalents to VL has been shown in vitro and in human skin ex vitro to generate ROS leading to induction of pro-inflammatory cytokines and MMP-1 and MMP-9[11]. Dose-dependent generation of hydrogen peroxide was also measured after exposure to VL. Certainly this oxidative insult, the inflammatory cascades, and the MMPs destroy dermal matrix to contribute to extrinsic photoaging.
IR solar radiation
Almost 50% of the solar energy reaching the earth’s surface is IR or heat energy: IR-A (λ = 700-1400 nm), IR-B (λ = 1400-3000 nm), and IR-C (λ = 3000 nm - 1 mm). Solar terrestrial radiation is about 30% IR-A. IR-B and IR-C do not penetrate deeply into the skin, but they do contribute to heating the skin. Direct sunlight can raise the temperature of human skin from 37 °C to 40 °C, with darker skin types IV-VI responding to IR exposure with greater rises in temperature than experienced by light-skinned individuals (types I and II). Although more than 65% of incipient IR-A penetrates to the dermis and 10% to the subcutaneous fat, normal (non-excessive) exposure does not raise the skin’s temperature[12].
The reaction to IR-A varies with skin type: In darker skin, melanin synthesis is stimulated with little effect in dermal MMPs, while in lightly pigmented skin, collagen in the dermal extracellular matrix is altered not only by destruction through activation of MMPs, but also by a direct reduction in synthesis[12]. Thus lightly pigmented skin manifests wrinkles and a crepe-like quality, while darkly pigmented skin responds with increased solar lentigos - both suffering from premature extrinsic aging. In addition, chronic exposure to IR-A induces angiogenesis and unattractive erythema ab igne as seen in “bakers’ arms” and “glassblowers’ faces”[12].
Although the clinical manifestations of photoaging of the skin are similar after UV and IR-A exposure, the mechanisms of damage are different. IR-A affects the mitochondrial electron transport chain, increasing mitochondrial superoxide anion production[13]. In human fibroblasts in vitro, IR-A induces genes regulating apoptosis, extracellular matrix, calcium regulation, and other signaling[14]. Solar elastosis is markedly exacerbated by IR-A in two ways: (1) stimulation of MMP-12 which specifically degrades elastin; and (2) abnormal production of the major components of elastin-fibrillin-1,2 and tropoelastin[13]. Along with degradation of collagen, this accumulation of abnormal, clumped elastin in the dermis is the paramount cause of the crepe-like, saggy, wrinkled appearance of photoaged skin.
Synergy: UVA and pollutants
Atmospheric pollutants fill the air in modern cities. Outside, traffic exhausts, power plants and factories pollute, and, inside, stoves, heaters, fireplaces, candles, and cigarette smoke. Organic and inorganic compounds are emitted. PAHs are ubiquitous in our air, water, and food. The most significant PAH in urban environments is benzo[a]pyrene (BaP). These PAHs are absorbed onto and within the larger PM pollutants. When inhaled or absorbed after direct contact with mucosa or skin, these PAHs are metabolized to quinones which generate ROS.
Today, the levels of PAHs are lower than those of 40-50 years ago, well below a toxic or carcinogenic level. However, with simultaneous exposure of the skin to PAHs and UVA radiation, synergistic interactions increase oxidative damage, leading to increased extrinsic damage to the skin with resultant photoaging and skin cancer.
In mouse model experiments, exposure to low doses of BaP alone, or to UVA alone, does not initiate skin cancer. However, exposure to 1.8% of the carcinogenic dose of BαP with simultaneous exposure to 12.5% of the carcinogenic dose of UVA could initiate skin cancer[1,15], as shown in Figure 2 (this exposure to UVA is comparable to 2 h of midday sunlight on a summer day in New York City).
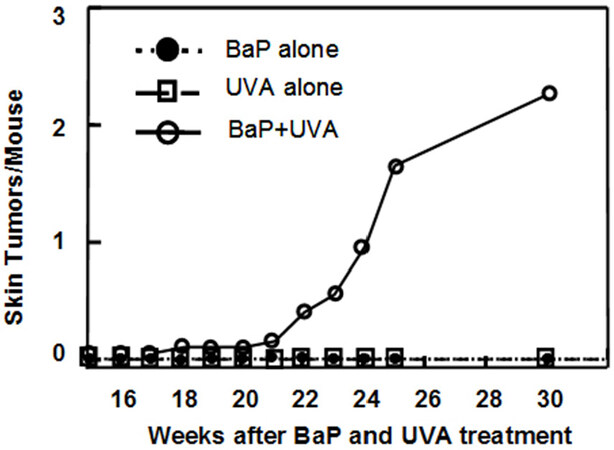
Figure 2. Squamous cell carcinoma multiplicity (number of tumors/mouse) in Skh:1 female mice after topical application of BaP (8 nmol) followed by exposure to ultraviolet A (UVA) (40 kg/m2) three times weekly for 25 weeks. For 5 weeks after treatments were discontinued, tumors were still counted (Wang et al.[1]). (Printed with consent of the authors.). BaP: benzo[a]pyrene
Synergistic damage was further demonstrated in vitro. High concentrations of d-OHdG and hydrogen peroxide (H2O2) were generated when calf-thymus DNA was exposed to UVA after incubation with BaP; in contrast, only minimal concentrations were measured when exposed to UVB after BaP[16]. Cells exposed to UVA after having been incubated with BaP increased 8-OHdG by a factor of 17, while exposure to UVB resulted in only an increase by a factor of 3[1]. Another experiment exposed human keratinocytes to UVA after incubation with BαP: H2O2 production increased 6-fold compared to only a factor of 1.2 after exposure to UVB[1]. In the skin, all of this oxidative damage manifests clinically as photoaging and proclivity to carcinogenesis.
With exposure to UV, BaP forms diol epoxide (BPDE) which combines covalently to DNA to form BPDE-DNA adducts that directly initiate carcinogenesis[17]. Exposure to UVA generates more than double the number generated by exposure to UVB[18]. These adducts directly produce ROS. Furthermore, the BaP is recycled within cells, a reaction enhanced by UVA exposure, making even small amounts of BaP extremely damaging. This sequence of reactions leading to photoaging of the epidermis and dermis as well as to initiation and promotion of skin cancer is shown in Figure 3.
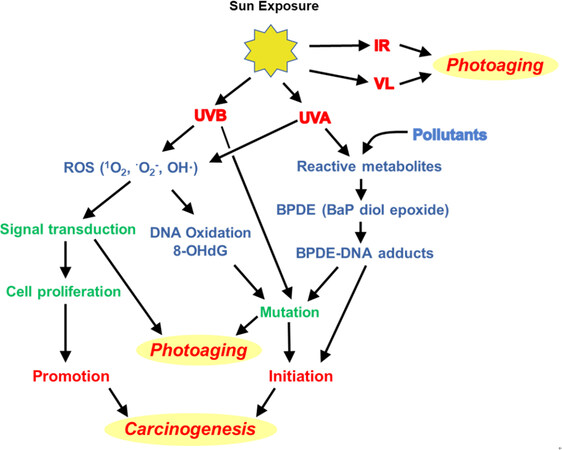
Figure 3. Mechanisms of solar-induced photoaging and carcinogenesis of the skin. Solar ultraviolet B (UVB) directly mutates cellular DNA to cause photoaging and to initiate carcinogenesis. Ultraviolet A (UVA) interacts synergistically with pollutants to form DNA-adducts, initiating carcinogenesis. Both UVA and UVB generate ROS that oxidize DNA to form 8-OHdG, leading directly to photoaging and tumor initiation. ROSs further activate signal transduction of inflammatory cytokines and matrix-destructive enzymes, which exacerbate photoaging and cellular proliferation to promote carcinogenesis. Exposure to solar (and technological) VL and IR heat also induces photoaging of the skin. (Modified from Saladi et al.[18] with consent of authors.). ROS: reactive oxygen species; 8-OHdG: 8-hydroxy deoxyguanosine; VL: visible light; IR: infrared light
Ozone
In the stratosphere (10-30 miles above the earth’s surface), O3 protects us by blocking dangerous solar UVC and dangerous high-energy UVB. On the other hand, low level tropospheric O3 causes significant oxidative damage, particularly to the skin. The average level of O3 on the earth’s surface is about 0.05-0.1 ppm, quite low in comparison with the stratosphere with concentrations of 10 ppm. However, especially during the summer, in densely populated cities such as Mexico City (with the highest levels) and even Rome and Paris, industry and traffic emit VOCs including nitrogen oxides, methane, carbon monoxide, and sulfuric compounds which accumulate, causing a progressive increase in O3 concentrations up to 0.8 ppm.
Although O3 cannot penetrate the skin, it oxidizes lipids on the skin’s surface, thereby triggering destructive inflammatory cascades in deeper cellular layers, turning on genes to produce inflammatory cytokines - interleukin-8 (Iℓ-8), tumor necrosis factor-alpha (TNF-α), transforming growth factor beta (TGF-β), cyclooxygenase-2 (COX-2), intercellular adhesion molecule (ICAM), and vascular cell adhesion molecule (VCAM)[19,20] - all of which have been measured in response to surface O3. This inflammatory onslaught certainly disrupts dermal cells and extracellular matrix to exacerbate photoaging[21]. Further damage is rendered by O3 generation of the protein adduct 4-hydroxy-2-nonenal (HNE) and carbonyl protein adducts[22].
Direct proof that O3 exposure contributes substantially to accelerating extrinsic aging was demonstrated in Skh:1 hairless mice[22,23]. With exposure to O3, dermal MMP-2 (a gelatinase that digests collagen I and IV) and MMP-12 (an elastase) are activated; MMP-9 (a gelatinase with activity similar to MMP-2) is activated only in older mice[23]. O3 further inhibits activation of inhibitors of MMPs[24], thus increasing matrix degradation with resultant manifestations of dermal aging: Wrinkles, crepe-like skin, and sagging due to loss of support, seen histologically as loss of collagen with disorganization of fibrillar alignment and loss of elastin with clumping of dysfunctional elastic tissue fragments. This degradation of firm extracellular matrix allows and promotes enhanced tumor growth[25].
Defending against this damage are antioxidants vitamin C and vitamin E on the skin’s surface. However, exposure to O3 rapidly depletes these essential antioxidants[26], probably because the resident levels of these vitamins are rapidly depleted by quenching the initial onslaught of O3-induced oxidative damage. Thus, the first antioxidant defense is lost. Another protective mechanism is the induction of epidermal heat shock proteins (HSP) sequentially: HSP 27 (at 2 h), HSP 70 (at 12 h), and HSP 32 (maximally at 24 h). HSP 27 actually increases 20-fold and HSP 70 by 8-fold in response to O3. These HSPs may mitigate the oxidative harm from surface O3[19]. Further defending against all of the O3-induced oxidative stress is activation of the antioxidant transcription factor nuclear factor of kappa-light-chain-enhancers of activated B-cells (NF-κB) in keratinocytes as well as in dermal cells, as demonstrated in vitro and in vivo[19,22].
A recently published epidemiological study from two cohort groups in Germany (of 2013 Caucasian men and women)[27] provides direct proof that exposure to tropospheric O3 does indeed exacerbate extrinsic skin aging. Five-year cumulative residential exposure to ambient O3 was calculated for each of the two neighborhoods (within a 5-digit postal code). Course wrinkles and hyperpigmentation (solar lentigines) on the forehead, under eyes, crow’s feet area, and upper lip and facial lentigines were quantitated by visual scoring according to the validated Score of Intrinsic and Extrinsic Skin Aging (SCINEXATM)[27]. Correlation of course facial wrinkles with cumulative neighborhood O3 exposure was confirmed, but increased pigmented lentigines were not noted. This correlation was independent of other possible environmental confounders and airborne pollutants including particulate matter.
Particulate matter
Even the earliest humans suffered from particulate matter exposure: Dust storms from the Sahara, and pollen and particulate fecal matter from grazing herds on the savannah caused respiratory, cardiovascular, and skin disease. Today, city environments are filled with larger PM10 and PM2.5 particles (≤ 10 μm and ≤ 2.5 μm diameter, respectively) of soot, primarily emitted by diesel engines, factories, power plants, and incinerators as well as smaller ultrafine particles (UFPs) (≤ 100 nm, 0.1 μm, diameter) from traffic. Up to 475 kg/km2 are spewed into urban air each year[28]. Even rural dwellers suffer from nearby highways and inside furnaces and fireplaces as well as from forest fires and wind-blown dust. Polluting compounds (PAHs, NO2, SO2, and trace minerals) cover the surfaces and are incorporated into the cores of the PMs[29,30]. These PMs contacting skin are absorbed to the mid-stratum corneum, as shown by tape-stripping chimney sweeps[31]. UFPs not only penetrate the skin transdermally and through hair follicles[32,33], but also are inhaled through the lung to enter pulmonary circulation; PM contaminants can be measured in the blood within one hour and can remain for weeks[34].
Direct damage to the skin by larger PMs was shown by exposing reconstituted human epidermis in vitro to concentrated PM2.5[35]. Oxidation of surface lipids and apoptosis correlated with increasing dose and exposure time. The impact of larger PM10 pollution has been demonstrated by in vitro exposure of human dermal fibroblasts to PM10 for 24 h[36]. Surprisingly, 1,977 genes were expressed, most significantly pro-inflammatory genes for interleukins (Iℓ) Iℓ-1β, Iℓ-6, Iℓ-8, and Iℓ-33, resulting in increases especially in Iℓ-6 and -8, cytochrome (CYP) P450 (CYP1A1, CYP1B1), and MMP-1 and -3, and accompanied by substantial decreases in transforming growth factor-β (TGF-β) and in collagen I and elastin mRNA. All of these factors directly result in the crepe-like, wrinkled, and sagging quality of extrinsic aging.
The mechanism of PM cutaneous insult is by oxidation of surface lipids with resultant ROS that activate inflammation through transcription factors such as NF-κB, turning on gene transcription for cytokines and Iℓ-1α, Iℓ-8, COX-1 and -2[37,38]. In particular, PMs containing quinones or trace metals localize in keratinocyte mitochondria, altering the mitochondrial ultrastructure with first dilation and vacuolization and loss of cristae, then thickening and shrinking[39].
VOCs
VOCs such as car exhaust with benzene and industrial emissions (mainly tetrachloroethylene) pollute the outdoor environment, while organic solvents in paints, varnishes, refinishing chemicals, glues, cleaning agents, cosmetics (especially nail polish) such as aliphatic hydrocarbons (formaldehyde and acetone) fill indoor closed spaces. Indoor pollution is particularly exacerbated in all homes in winter when windows are kept closed, and throughout the year in cities where skyscrapers and other buildings have sealed windows to maintain energy conservation. This VOC-contaminated air is recirculated, leading to “sick building syndrome.” Workers experience conjunctivitis, rhinitis, atopic dermatitis, eczema, and other contact or irritant dermatitis, particularly after reconstruction, painting, installation of new rugs and draperies, and after thorough cleaning. These airborne irritants contribute to extrinsic aging of exposed skin, particularly on the hands, neck, and face.
Tobacco smoke
A major indoor pollutant is cigarette (and cigar and pipe) smoke which exposes not only the smoker but also others to the “sidestream” smoke. In the closed environments of our modern world - with urban skyscrapers and airplanes and mass transit of subways - the VOCs and tobacco contaminants linger. In a cinema which had been designated “nonsmoking” for over 5 years, 35 tobacco-related chemicals were measured, carried inside by smokers’ hair, skin, and clothes - leading to “third-hand exposure”[40].
More than 4,700 different chemicals are released by burning tobacco[41], some as PM (including nicotine, quinone, and benzopyrenes), some as VOCs (PAHs, CO2, CO, formaldehyde, nitrosamine and many others). One dominant component of tobacco smoke, 4-aminobiphenyl, is particularly dangerous because it forms adducts on hemoglobin, albumin, and collagen[42]. These adducts remain for the “lifetime” of each component: On hemoglobin, adducts remain for 120 days at levels 5.5 times higher in smokers than nonsmokers; on albumin, adducts remain for 30 days, and on collagen, for up to 10 months. Oxygen delivery is impeded and collagen structure is disrupted, leading to extrinsic aging of the skin.
The long-term exposure of mice to cigarette smoke and UV results in barrier disruption, with transepidermal water loss, erythema, telangiectasis, decreased elasticity, visible crepe-like skin, and an increased incidence of benign epitheliomas and squamous cell carcinomas[43]. Increased mRNA for the metalloproteinases MMP-1 and -3 with consequent breakdown of collagen I and III[44] as well as of elastic tissue is seen, accelerating extrinsic aging.
Mechanism of detoxification: the AHR
AHR evolved to detoxify food and airborne toxins. The AHR is found on cells of the gastrointestinal tract, pulmonary epithelium, and is especially highly expressed on all cells of the skin - keratinocytes, fibroblasts, and melanocytes as well as on regulatory T-cells and dendritic cells. The AHR recognizes and binds xenobiotic pollutants, then signals gene transcription of enzymes and inflammatory cytokines to metabolize these foreign substances[45]. The AHR plays a key role in barrier function, pigmentation regulation responding to environmental onslaughts, and skin inflammation[46]. Specific ligands activate specific genes. In the epidermis, UVB is absorbed by the amino acid tryptophan to form the photoproduct 6-formylindolo[3,2-b]carbazole (FICZ) and indoles which bind with high affinity to the AHR to induce the protective signaling cascade. Other potent ligands are the PAHs of tobacco smoke and city pollution as well as pesticide components such as tetrachlorodibenzo-p-dioxin (TCDD), the contaminant of Agent Orange, and even normal skin flora such as bacteria (non-pathogenic as well as pathogenic staphylococci) and yeasts (Malassezia furfur).
As seen in Figure 4, the inactive AHR is incorporated into a multi-protein complex in the cellular cytoplasm. Binding of the pollutant ligand generates a conformational change that breaks this complex so that ligand-AHR enters the nucleus where it dimerizes with AHR nuclear translocator (ARNT). This dimer is a transcription factor which binds to genes to induce DNA transcription. AHR can selectively induce transcription of NF-κB, leading to inflammatory cytokines such as NRF2 (which activates antioxidants) as well as to cell cycle (proliferation) and immune modulators.
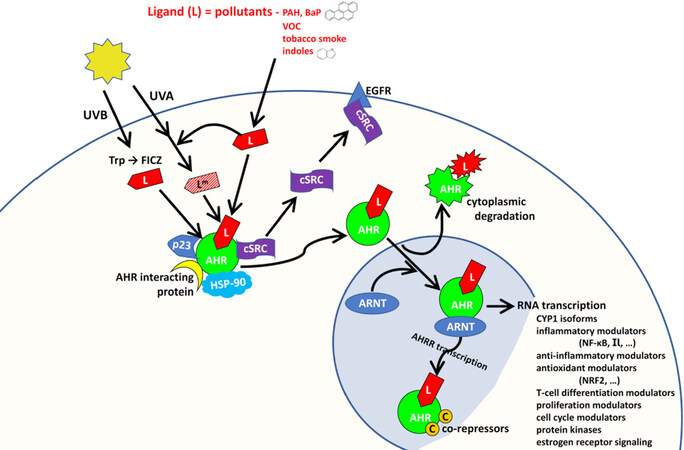
Figure 4. The aryl hydrocarbon receptor (AHR). The AHR is found in the cytoplasm bound to protein chaperone molecules, including AHR interacting protein, heat shock protein-90, the p53 protein, and the tyrosine kinase oncogene cSRC. When a pollutant ligand binds to this complex, conformational changes dissemble the complex so that the ligand-AHR unit translocates into the nucleus where it dimerizes with ARNT. The ligand-AHR-ARNT entity induces transcription of target genes, specific for each xenobiotic ligand. Overstimulation by the ligand-AHR complex is prevented (1) by RNA transcription of the AHR repressor (AHRR) gene to synthesize co-repressors; and (2) by cytoplasmic degradation of the ligand-AHR entity after expulsion from the nucleus. After release from the initial cytoplasmic AHR complex, the chaperone protein oncogene cSRC binds to the epidermal growth factor receptor (EGFR), activating the cSRC protein kinase to induce carcinogenesis. (Modified from Figure 1 of Vogeley et al.[47] with the author’s consent.). cSRC: “cellular sarcoma”, photo oncogene tyrosine-protein kinase; ARNT: AHR nuclear translocator
Alternatively, the ligand-AHR can induce the AHRR gene which inactivates the AHR by transcribing co-repressors[47]. Other inhibition of AHR is regulated by expulsion of the ligand-AHR from the nucleus for degradation in the cytoplasm. Thus overstimulation by the pollutant-ligand-AHR complex is curtailed. The co-chaperones that are released by the dissociation of the initial cytoplasm AHR complex after ligand binding lead to important cellular reactions: The heat shock proteins which are co-chaperoned by p23 stabilize or refold proteins that act to mitigate cellular stress; p23 also binds to the progesterone receptor that transports progesterone to the nucleus; the oncogene cSRC binds to the EGFR leading to activation of cSRC kinase and carcinogenesis.
Epidemiological correlation of extrinsic skin aging and pollution
Outdoor pollution
Vierkötter et al.[48] published an impressive epidemiological study showing the impact of urban pollution on aging skin on four-hundred Caucasian women (70-80 years old) who had previously enlisted in a study of the effect of industrial pollution on pulmonary aging. Half lived in urban Rohr and half in rural Bergen. All were evaluated by the SCINEXA scale which distinguishes intrinsic from extrinsic aging. Childhood sun exposure and smoking history were evaluated for each patient, and exposure to traffic and industrial emission at each individual’s home were measured. The researchers concluded that traffic-generated PM nanoparticles produced 16% more lentigines on the forehead and 17% more on the cheeks for each quartile increase in pollution measured. Concentrated soot increased lentigines by 27% on the forehead and 20% on the cheeks. Women living within 100 m of a busy road showed an increase of 25% and 20% in forehead and cheeks lentigines, respectively, for each interquartile increase in pollution.
A further study looked at the VOC NO2 released by traffic, known to cause lung damage and lung cancer[49]. Facial cheek lentigines were evaluated in women over 50 years old - 806 European Caucasians and 1,072 Chinese, making this the largest study to date of traffic-induced lentigines. Indeed, NO2 exposure increased lentigines in both cohorts. Lentigines develop at a younger age in Asians despite their cultural avoidance of sun exposure (in contrast to many Caucasians). This proclivity for lentigines may be due to a particular genetic marker in the SLC45A2 gene similarly noted in Japanese, a marker known to be involved in melanin synthesis[50].
Indoor pollution
Cigarette smoke is the most significant indoor contaminant; second-hand side-stream smoke permeates indoor space and is recycled in closed environment; third-hand smoke is carried by the hair, skin, and clothes of smokers entering a closed space[40]. In 1969, Henry Daniell recognized that smokers look older than non-smokers[51]. Several epidemiological papers confirm this observation[52,53,54]. Analysis shows that men who smoke have 2.3 times as many wrinkles as nonsmoking males, and women have 3.1 times as many as non-smoking women[53]. Smokers’ skin displays extensive aging with a leathery texture, dryness, deep wrinkles, a crepe-like quality, and sagging. This appearance of prematurely aged skin can be clearly seen in Figure 5. The 52-year-old smoker has far more severe periorbital wrinkles than her non-smoking 57-year-old cousin and neighbor[55]. A further dramatic demonstration of the ravages of photoaging in smokers was shown in photographs of identical twins with a greater than 5-year difference in smoking[56]. Especially upper eyelid skin redundancy, lower lid bags, molar bags, nasolabial folds, upper and lower lip wrinkles, and jowls are markedly worse in the twin who smoked.
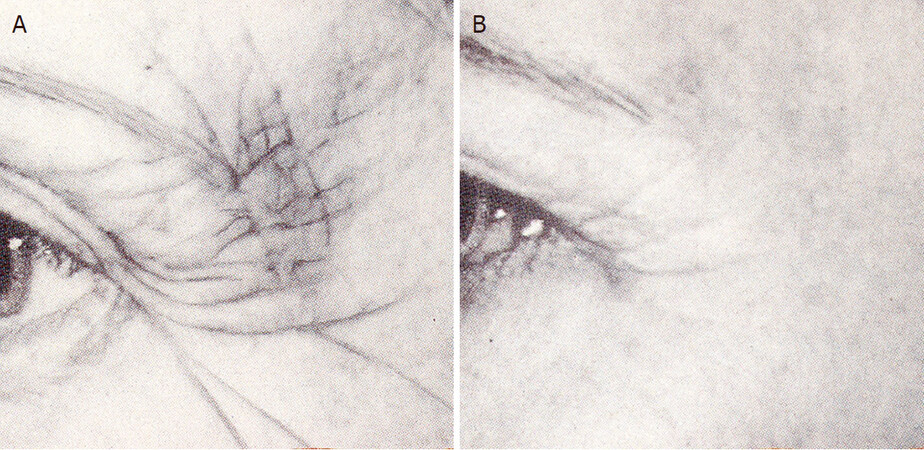
Figure 5. The 52-year old smoker (A) has more severe, deeper periorbital wrinkles than her 57-year-old non-smoking cousin (B). These cousins are neighbors who experienced the same environmental conditions throughout their lives (reprinted from Great Skin for Life, with consent of the author)
The other primary sources of indoor pollution, especially in underdeveloped countries, is cooking and heating with coal or wood. In China, more than 20% and up to 70% of households cook with solid fuel[57], causing chronic obstructive pulmonary disease, compromised lung function, and lung cancer[58]. To study the effect on the appearance of the skin, Li et al.[59] compared 405 women (of age 30 to 70 years) from northern China with 857 women from southern China, using the standardized SCINEXA evaluation. Correcting for age, cooking and heating with solid fuel significantly increased severe facial wrinkling by 5%-8% and by 75% the fine wrinkles of the hands.
Not only do underdeveloped countries suffer with indoor pollutants, but also throughout the world, gas stoves spew pollutants (primarily nitrogen dioxide and carbon monoxide) which make indoor air factors dirtier than outdoors, as reported by The Guardian (United Kingdom) recently. Candles and fireplaces further contribute. No specific studies on the effect of these frequent enjoyments on aging skin have been completed as yet, but certainly we know that these airborne contaminants are detrimental to skin.
Skin cancer
The most severe and devastating manifestation of photoaging is skin cancer. Here there is no doubt that environmental pollution directly initiates and propagates skin cancer. The first realization of environmental pollutants causing skin damage was in London in 1775 when Dr. Percivall Pott correlated SCC on the underside of the scrotum after exposure to soot in young boy chimney sweeps[60].
That UVB directly initiates all types of skin cancer - SCC, BCC, and MM - has been proven beyond doubt. UVA promotes skin cancer by generating specific DNA mutations, by production of labile ROS and subsequent oxidation of DNA, and by suppressing the immune response. As described above and shown in Figure 2, UVA also interacts synergistically with environmental airborne pollutants to form mutagenic DNA adducts leading to skin carcinogenesis.
The highly complex regulation of cellular physiology by the AHR and its influence on the initiation and propagation of skin cancer has been excellently reviewed by Vogeley et al.[47]. Some downstream reactions initiated by AHR suppress or inhibit initiation and promotion of carcinogenesis; others stimulate carcinogenesis. The regulation is highly specific for each xenobiotic pollutant, and concentration and interaction with UVA certainly contribute.
UVB invokes the AHR by generating the photoproducts, most prominently FICZ. The AHR-FICA complex initiates rapid metabolism of FICZ to limit carcinogenesis. On the other hand, high levels of UVB trigger other cytokines which stimulate SCC growth. Nitrosamines and aromatic amines from tobacco smoke and high levels of PAHs encountered with high industrial exposure activate the AHR, often leading to downstream metabolism of these xenobiotics to genotoxins within keratinocytes. AHR activation by these pollutants in Langerhans cells changes these antigen-presenting dendritic cells from stimulatory to regulatory, enhancing immunosuppression to allow tumor growth. Vietnam veterans had an increased incidence of melanoma after exposure to the Agent Orange carcinogen TCDD[61], which through the AHR-TCDD complex initiated reactions that alter the microenvironment by activating destructive MMPs[62], inducing angiogenesis, and increasing cancer cell motility[63].
Perhaps the most studied clinical correlation of skin (and lung) cancer to environmental pollutants is the correlation to cigarette smoking. Smokers statistically have twice as many melanomas as non-smokers, 1.5 times as many cutaneous SCCs, 15 times as many SCCs of the lip, and 78 times as many oral epithelial cancers. Another direct chemical correlation of the incidence of mortality of skin cancer induced by environmental exposure was studied in São Paulo, Brazil[64]. Indeed, higher PM10 pollution is directly associated with an increased incidence of skin cancer.
Conclusion
The appearance of aging on our skin is primarily caused by exposure to solar radiation and environmental airborne pollutants. Recent research has elucidated new insights into the molecular mechanisms of skin damage and natural defenses. We now realize that not only is UVB the main instigator of photoaging, but also UVA as well as visible light and long wavelength IR heat contribute to extrinsic aging. Environmental airborne pollutants - particularly the PAHs released by industry and traffic, O3, VOCs, PM coated with and containing xenochemicals - all directly cause the hallmark appearance of premature aging of the skin. Furthermore, exposure to UVA and airborne pollutants simultaneously causes synergistic damage and accelerated extrinsic aging with increased carcinogenesis.
The new epidemiologically-proven realizations about the specific causes of damage can teach how to prevent or lessen the adverse results of exposure. Avoidance of sun exposure and use of broad-spectrum sunscreen; improvements in technology to minimize industrial pollution and decreasing traffic emissions; stopping smoking; avoiding cooking and heating with fossil fuels in closed, indoor spaces; choosing rural over urban dwellings if possible - these all reduce the risk. In Korea, indoor air quality was improved in nine kindergarten classes, decreasing PM10 significantly from 182.7 μg/m to 73.4 μg/m[28]. The European Health Event project improved indoor air quality by optimizing ventilation, filtering outdoor air, and controlling indoor sources of pollution (especially by prohibiting smoking)[65]. Industry is developing technology to be ecofriendly - for example, the use of compressed natural gas as fuel instead of coal and petroleum decreases PM pollution.
All environmental exposure - whether from solar energy or airborne toxins - leads to skin damage by generation of ROS, either directly or via the AHR receptor. Our bodies’ primary protective mechanism is our complex endogenous antioxidant network which is dependent upon external supplementation - either orally, topically, or both. These antioxidant-induced reactions have been shown to combat extrinsic aging of the skin and skin cancer. Indeed, the resident epidermal antioxidants vitamin C and vitamin E are depleted 55% and 25%, respectively, as they protect surface skin after exposure to high concentrations of O3[26].
Antioxidants can best be replenished or delivered to the skin by topical application giving far higher concentrations than attained by oral ingestion. For example, L-ascorbic acid (vitamin C) 15% gives 27-40 times the skin concentration resulting from high oral intake[66], d-α-tocopherol 5% (the only one of 32 forms of vitamin E that is effective on skin), by a factor of 12[67], and selenium (L-selenomethionine 0.05%), by a factor of 8[68]. These three antioxidants have been extensively studied and proven to prevent and even reverse photoaging of the skin in mice[67,68,69], pigs[70], and humans[71,72], and to inhibit UV-induced skin cancer in mice[67,68,69,73]. However, stringent criteria are required for topical formulations to be stable, to be successfully absorbed, as to be active as antioxidants. Esterified forms of vitamins C[66] and E[67] are not absorbed transcutaneously, and the ester is not reduced to the -OH form required for antioxidant activity. Only the isomer d-α-tocopherol is effective; the other 31 isomers (eight “dl” isomer configurations and four α, β, γ, δ forms) are not[67]. High concentrations are required, optimally 15%-20% for l-ascorbic acid[72] and 2%-5% for d-α-tocopherol[67]. Biologic L-selenomethionine is the optimal form of the multiple valence forms of selenium (at concentrations of 0.02%-0.05%)[68]. Combining L-ascorbic acid (15%) with α-tocopherol (1%) gives four-fold protection against clinical minimal erythema dose (MED) and against thiamine dimer formation alone[74]; addition of another plant antioxidant ferulic acid (0.5%) yields 8-fold protection[72]. This formulation was shown to prevent the up-regulation of oxidative and inflammatory markers in human skin explants exposed to UV plus O3 and diesel engine exhaust[75]. Many other topical antioxidants are being actively studied, including lycopene[76], genistein[77], plant-derived phenolic compounds (such as green tea, pomegranate, grape, and cocoa), and certain marine algae[78].
Current research is also focused on understanding at a cellular level how the AHR binds and metabolizes xenochemicals, leading to complex cascades of protective or damaging inflammatory reactions, depending upon the specific pollutant, simultaneous exposure to UVA and to other pollutants, and concentration of these foreign substances. This understanding may lead to the discovery of methods to modulate the AHR response in order to prevent and protect the skin from extrinsic damage.
Declarations
AcknowledgementsThe author would like to thank Xueyan Zhou, MD, MS, for assistance with literature research and Heather Nolan, MA, for excellent artistic rendition of the figures, literature research, and editing of text.
Authors’ contributionsThe sole author wrote this review article.
Availability of data and materialsNot applicable.
Financial support and sponsorshipNone.
Conflicts of interestThe author declared that there are no conflicts of interest.
Ethical approval and consent to participateNot applicable.
Consent for publicationSubjects shown in Figures 1 and 5 did give consent.
Copyright© The Author(s) 2020.
REFERENCES
1. Wang Y, Saladi R, Wei H. Synergistic carcinogenesis of chemical carcinogens and long wave-length UVA radiation. Trends Photochem Photobio 2001;10:31-45.
2. Gilchrest BA, Blog FB, Szabo G. Effects of aging and chronic sun exposure on melanocytes in human skin. J Invest Dermatol 1979;73:141-3.
3. Bolognia JL, Orlow SJ. Chapter 65 - Melanocyte Biology. In: Bolognia JL, Schaffer JV, Cerroni L, editors. Dermatology Fourth Edition. New York: Elsevier; 2018. pp. 1075-86.
4. Premi S, Wallisch S, Mano CM, Weiner AB, Bacchiocchi A, et al. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015;347:842-7.
5. Lagarrigue SG, George J, Questel E, Lauze C, Meyer N, et al. In vivo quantification of epidermis pigmentation and dermis papilla density with reflectance confocal microscopy: variations with age and skin phototype. Exp Dermatol 2012;21:281-6.
6. Cadet J, Douki T. Formation of UV-induced DNA damage contributing to skin cancer development. Photochem Photobiol Sci 2018;17:1816-41.
7. Kollias N, Baqer A. An experimental study of the changes in pigmentation in human skin
8. Rosen CF, Jacques SL, Stuart ME, Gange RW. Immediate pigment darkening: visual and reflectance spectrophotometric analysis of action spectrum. Photochem Photobiol 1990;51:583-8.
9. Pathak MA, Riley FC, Fitzpatrick TB. Melanogenesis in human skin following exposure to long-wave ultraviolet and visible light. J Invest Dermatol 1962;39:435-43.
10. Mahmoud BH, Ruvolo E, Hexsel CL, Liu Y, Owen MR, et al. Impact of long-wavelength UVA and visible light on melanocompetent skin. J Invest Dermatol 2010;130:2092-7.
11. Liebel F, Kaur S, Ruvolo E, Kollias N, Southall MD. Irradiation of skin with visible light induces reactive oxygen species and matrix-degrading enzymes. J Invest Dermatol 2012;132:1901-7.
12. Cho S, Shin MH, Kim YK, Seo JE, Lee YM, et al. Effects of infrared radiation and heat on human skin aging
13. Schroeder P, Calles C, Benesova T, Macaluso F, Krutmann J. Photoprotection beyond ultraviolet radiation--effective sun protection has to include protection against infrared a radiation-induced skin damage. Skin Pharmacol Physiol 2010;23:15-7.
14. Calles C, Schneider M, Macaluso F, Benesova T, Krutmann J, et al. Infrared a radiation influences the skin fibroblast transcriptome: mechanisms and consequences. J Invest Dermatol 2010;130:1524-36.
15. Kelfkens G, de Gruijl FR, van der Leun JC. Tumorigenesis by short-wave ultraviolet A: papillomas versus squamous cell carcinomas. Carcinogenesis 1991;12:1377-82.
16. Shyong EQ, Lu Y, Goldstein A, Lebwohl M, Wei H. Synergistic enhancement of H2O2 production in human epidermoid carcinoma cells by benzo[a]pyrene and ultraviolet A radiation. Toxicol Appl Pharmacol 2003;188:104-9.
17. Casale GP, Singhai M, Bhattacharya S, RamaNathan R, Roberts KP, et al. Detection and quantification of depurinated Benzo[a]pyrene-adducted DNA bases in the urine of cigarette smokers and women exposed to household coal smoke. Chem Res Toxicol 2001;14:192-201.
18. Saladi R, Austin L, Gao D, Lu Y, Phelps R, et al. The combination of benzo[a]pyrene and ultraviolet A causes an in vivo time-related accumulation of DNA damage in mouse skin. Photochem Photobio 2003;77:413-9.
20. Valacchi G, Sticozzi C, Belmonte G, Cervellati F, Demaude F, et al. Vitamin C compound mixtures prevent ozone-induced oxidative damage in human keratinocytes as initial assessment of pollution protection. PLoS One 2015;10:e0131097.
21. Pittayapruek P, Meephansan J, Prapapan O, Komine M, Ohtsuki M. Role of matrix metalloproteinases in photoaging and photocarcinogenesis. Int J Mol Sci 2016;17:868.
22. Valacchi G, Pagnin E, Okamoto T, Corbacho AM, Olano E, et al. Induction of stress proteins and MMP-9 by 0.8 ppm of ozone in murine skin. Biochem Biophys Res Commun 2003;305:741-6.
23. Fortino V, Maioli E, Torricelli C, Davis P, Valacchi G. Cutaneous MMPs are differently modulated by environmental stressors in old and young mice. Toxicol Lett 2007;173:73-9.
24. Caley MP, Martins VLC, O’Toole EA. Metalloproteinases and wound healing. Adv Wound Care 2015;4:225-34.
25. Xu X, Wang Y, Chen Z, Sternlicht MD, Hidalgo M, et al. Matrix metalloproteinase-2 contributes to cancer cell migration on collagen. Cancer Res 2005;65:130-36.
26. Thiele JJ, Traber MG, Tsang K, Cross CE, Packer L. In vivo exposure to ozone depletes vitamins C and E and induces lipid peroxidation in epidermal layers of murine skin. Free Radic Biol Med 1997;23:385-1.
27. Fuks KB, Hüls A, Sugiri D, Altug H, Vierkötter A, et al. Tropospheric ozone and skin aging: Results from two German cohort studies. Environ Int 2019;124:139-44.
28. Puri P, Nandar SK, Kathuria S, Ramesh V. Effects of air pollution on the skin: a review. Indian J Dermatol Venereol Leprol 2017;83:415-23.
29. Soeur J, Bolandi JP, Chollet C, Denat L, Dimitrov A, et al. Photo-pollution stress in skin. Traces of Pollutants (PAH and particulate matter) impair redox homeostasis in keratinocytes exposed to UVA1. J Dermatol Sci 2017;8:162-9.
30. Donaldson K, Tran L, Jimenez LA, Duffin R, Newby DE, et al. Combustion-derived nanoparticles: a review of their toxicology following inhalation exposure. Part Fibre Toxicol 2005;2:10-4.
31. Kammer R, Tinnerberg H, Briksson K. Evaluation of a tape-stripping technique for measuring dermal exposure to pyrene and benzo[a]pyrene. J Environ Monit 2011;13:2165-71.
32. Lademann J, Otberg N, Jacobi U, Hoffman RM, Blume-Peytavi U. Follicular penetration and targeting. J Invest Dermatol Symp Proc 2005;10:301-3.
33. Jin SP, Li Z, Choi EK, Lee S, Kim YK. Urban particulate matter in air pollution penetrates into the barrier-disrupted skin and produces ROS-dependent cutaneous inflammatory response
34. Moreau M, Ouellet N, Ayotte P, Bouchard M. Effects of intravenous benzo[a]pyrene dose administration on levels of exposure biomarkers, DNA adducts, and gene expression in rats. J Toxicol Environ Health A 2018;78:166-84.
35. Magnani ND, Muresan XM, Belmonte G, Cervellati F, Sticozzi C, et al. Skin damage mechanisms related to airborne particulate matter exposure. Toxicol Sci 2016;149:227-36.
36. Park SY, Byun EJ, Lee JD, Kim S, Kim HS. Air pollution, autophagy, and skin aging: Impact of particulate matter (PM10) on human dermal fibroblasts. Int J Molec Sci 2018;19:2727.
37. Marchini T, Magnani ND, Paz ML, Vanasco V, Tasat D, et al. Time course of systemic oxidative stress and inflammatory response induced by an acute exposure to Residual Oil Fly Ash. Toxicol and Applied Pharmacol 2014;274:274-82.
38. Cervellati F, Benedusi M, Manarini F, Woodby B, Russo M, et al. Proinflammatory properties and oxidative effects of atmospheric particle components in human keratinocytes. Chemosphere 2020;240:124746-53.
39. Zhang Y, Zheng L, Tuo J, Liu Q, Zhang X, et al. Analysis of PM2.5-induced cytotoxicity in human HaCaT cells based on a microfluidic system. Toxicol In Vitro 2017;43:1-8.
40. Sheu R, Stönner C, Ditto JC, Klüpfel T, Williams J, et al. Human transport of thirdhand tobacco smoke: A prominent source of hazardous air pollutants into indoor nonsmoking environments. Environ Stud 2020;6:eaay4109.
41. Valacchi G, Sticozzi C, Pecorelli A, Cervellati F, Cervellati C, et al. Cutaneous responses to environmental stressors. Ann NY Acad Sci 2012;1271:75-81.
42. Bryant MS, Vineis P, Skipper PL, Tannenbaum SR. Haemoglobin adducts of aromatic amines in people exposed to cigarette smoke. IARC Sci Publ 1998;89:133-136.
43. Pavlou P, Rallis M, Deliconstantinos G, Papaioannou G, Grando S. In-vivo data on the influence of tobacco smoke and UV light on murine skin. Toxicol and Indus Health 2009;25:231-9.
44. Yin L, Morita A, Tsuji T. Alternations of extracellular matrix induced by tobacco smoke extract. Arch Dermatol Res 2000;292:183-94.
45. Larigot L, Juricek L, Dairou J, Coumoul X. AhR signaling pathways and regulatory functions. Biochim Open 2018;7:1-9.
46. Haarmann-Stemmann T, Esser C, Krutman J. The Janus-faced role of aryl hydrocarbon receptor signaling in the skin: Consequences for prevention and treatment of skin disorders. J Invest Dermatol 2015;135:2572-6.
47. Vogeley C, Esser C, Tüting T, Krutmann J, Haarmann-Stemmann T. Role of the aryl hydrocarbon receptor in environmentally induced skin aging and skin carcinogenesis. Intl J Molec Sci 2019;20:6005-26.
48. Vierkötter A, Schikowski T, Ranft U, Sugiri D, Matsui M, et al. Airborne particle exposure and extrinsic aging. J Invest Dermatol 2010;130:2719-26.
49. Nakamura M, Morita A, Seité S, Haarmann-Stemmann T, Grether-Beck S, et al. Environment-induced lentigines: formation of solar lentigines beyond ultraviolet radiation. Exper Dermatol 2015;24:407-11.
50. Vierkötter A, Krämer U, Sugiri D, Morita A, Yamamoto A, et al. Development of lentigines in German and Japanese women correlate with variants in the SLC45A2 gene. J Invest Dermatol 2012;132:733-6.
52. Kadunce DP, Burr R, Gress R, Kanner R, Lyon JL, et al. Cigarette smoking: risk factor for premature facial wrinkling. Ann Intern Med 1991;114:840-4.
53. Ernster VL, Grady D, Miike R, Black D, Skelby J, et al. Facial wrinkling in men and women, by smoking status. Am J Public Health 1995;85:78-82.
54. Aizen E, Gilhar A. Smoking effect on skin wrinkling in the aged population. Int J Dermatol 2001;40:431-3.
55. Burke KE. Great Skin for Life. London: Hamlyn Books Ltd, An imprint of Reed International; 1996. p. 59.
56. Okada HC, Alleyne B, Varghai K, Kinder K, Guyuron B. Facial changes caused by smoking: A comparison between smoking and nonsmoking identical twins. Plast Reconstr Sugr 2013;132:1085-92.
57. Jin Y, Zhou Z, He G, Wei H, Liu J, et al. Geographical, spatial, and temporal distributions of multiple indoor air pollutants in four Chinese provinces. Environ Sci Technol 2005;39:8431-9.
58. Zhang JJ, Smith KR. Household air pollution from coal and biomass fuels in China: measurements, health impacts, and interventions. Environ Health Perspect 2007;115:848-35.
59. Li M, Vierkötter A, Schikowski T, Hüls A, Ding A, et al. Epidemiological evidence that indoor air pollution from cooking with solid fuels accelerates skin aging in Chinese women. J Dermatol Sci 2015;79:148-54.
60. Mukherjee S. The emperor of all maladies - a biography of cancer. New York: Scribner; 2010. p. 247.
61. Akhtar FZ, Garabrant DH, Ketchum NS, Michalek JE. Cancer in US air force veterans of the Vietnam War. J Occup Environ Med 2004;46:123-36.
62. Villano CM, Murphy KA, Akintobi A, White LA. 2,3,7,8-tetrachlorodibenzo-
63. Contador-Troca M, Alvarez-Barrientos A, Barrasa E, Rico-Leo EM, Catalina-Fernandez L, et al. The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carciogenesis 2013;34:2683-93.
64. Yanagi Y, Assunção JV, Barrozo LV. The impact of atmospheric particulate matter on cancer incidence and mortality in the city of São Paulo, Brazil. Cad Saude Publ 2012;28:1737-48.
65. Asikainen A, Carrer P, Kephalopoulous S, Fernandes E de O, Wargocki P, et al. Reducing burden of disease from residential indoor air exposures in Europe (HEALTHVENT project). Environ Health 2016;15 Suppl 1:35.
66. Pinnell SR, Yang HS, Omar M, Monteiro-Riviere N, DeBuys HV, et al. Topical L-ascorbic acid: percutaneous absorption studies. Dermatol Surg 2001;27:137-42.
67. Burke KE, Clive J, Combs GF Jr, Commisso J, Keen CL, et al. The effects of topical and oral vitamin E on pigmentation and skin cancer induced by ultraviolet irradiation in Skh:2 hairless mice. Nutr Cancer 2000;38:87-97.
68. Burke KE, Combs GF Jr, Gross EG, Bhuyan KC, Abu-Libdeh H. The effects of topical and oral L-selenomethionine on pigmentation and skin cancer induced by ultraviolet irradiation. Nutr Cancer 1992;17:123-37.
69. Burke KE. Method for the prevention and reversal of the extrinsic aging of the skin by transdermal application of selenoamino acids and compositions therefore. US Patent Number:; 1994.
70. Darr D, Combs S, Dunston S, Manning T, Pinnell S. Topical vitamin C protects porcine skin from ultraviolet radiation-induced damage. Br J Dermatol 1992;127:247-53.
71. Burke KE, Combs GF Jr, French IW, Skeffington D. The effect of topical L-selenomethionine on minimal erythema dose of ultraviolet irradiation in humans. Photoderm Photoimmun Photomed 1992;9:52-7.
72. Zielinski JE, Pinnell SR. Stabilized ascorbic acid compositions and methods thereof. US Patent; 2004.
73. Burke KE, Zhou X, Wang Y, Commisso J, Keen CL, et al. The effects of topical L-selenomethionine on protection against UVB-induced skin cancer when given before, during, and after UVB exposure. J Drugs Dermatol 2014;13:1214-23.
74. Lin JY, Selim A, Shea CR, Grichnik JM, Omar MM, et al. UV photoprotection by combination topical antioxidants vitamin C and E. J Am Acad Dermatol 2003;48:866-74.
75. Ferrara F, Woodby B, Pecorelli A, Schiavone ML, Pambianchi E, et al. Additive effect of combined pollutants to UV induced skin OxInflammation damage. Evaluating the protective topical application of a cosmeceutical mixture formulation. Redox Biol 2020;34:101481.
76. Zhou X, Burke KE, Wang Y, Wei H. Dietary lycopene protects SkH-1 mice against ultraviolet B-induced photocarcinogenesis. J Drugs Derm 2019;18:1214-23.
77. Burke KE. “Chapter 24: Photodamage: protection and reversal with topical antioxidants in Textbook of Cosmetic Dermatolgoy, Fifth Edition. Boca Raton, FL: Taylor and Francis; 2017. pp. 199-213.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Burke KE. Environmental aging of the skin: new insights. Plast Aesthet Res 2020;7:59. http://dx.doi.org/10.20517/2347-9264.2020.154
AMA Style
Burke KE. Environmental aging of the skin: new insights. Plastic and Aesthetic Research. 2020; 7: 59. http://dx.doi.org/10.20517/2347-9264.2020.154
Chicago/Turabian Style
Burke, Karen E.. 2020. "Environmental aging of the skin: new insights" Plastic and Aesthetic Research. 7: 59. http://dx.doi.org/10.20517/2347-9264.2020.154
ACS Style
Burke, KE. Environmental aging of the skin: new insights. Plast. Aesthet. Res. 2020, 7, 59. http://dx.doi.org/10.20517/2347-9264.2020.154
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 52 clicks
Cite This Article 52 clicks

 Like This Article 21
likes
Like This Article 21
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.