
A case of 25-year-old giant neurofibromatosis
 0
0Abstract
In this paper, the authors presented a case report of a 39-year-old man taken to the Emergency Unit with a 25-year-old giant neurofibromatosis. Moreover, the treatment applied for this case and the physiopathology of the neurofibromatosis disease are also discussed.
Keywords
Introduction
Neurofibromatosis is a neurocutaneous condition that can involve almost any organ system. Thus, the signs and symptoms presented may vary widely. Two major subtypes exist: neurofibromatosis 1 (NF-1), which is the most common subtype, referred to as peripheral NF, and neurofibromatosis 2 (NF-2), which is referred to as central NF.[1,2]
We report a case of a giant neurofibroma kept for 25 years without treatment.
Case report
A 39-year-old male patient arrived at the Emergency Department with a history of bleeding from a moll on his back for the last 3 days. The patient also sustained episodes of nausea with no associated vomit, weakness and sleepiness.
At examination, we found a giant mass on his sacoccocigeal region [Figures 1 and 2] with small areas of scarification with punctiform mild bleeding. Patient was mildly dispneic with taquicardia associated. No other symptoms were found. According to the patient, the mass started to develop at the age of 14 years and he never felt compelled to have that examined by doctor. In fact, he regarded this as a phenomenon rendering him different from others.
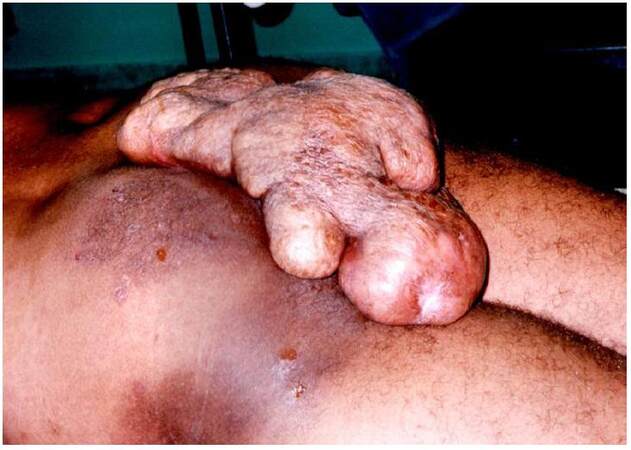
Figure 1. Lateral aspect of the tumor
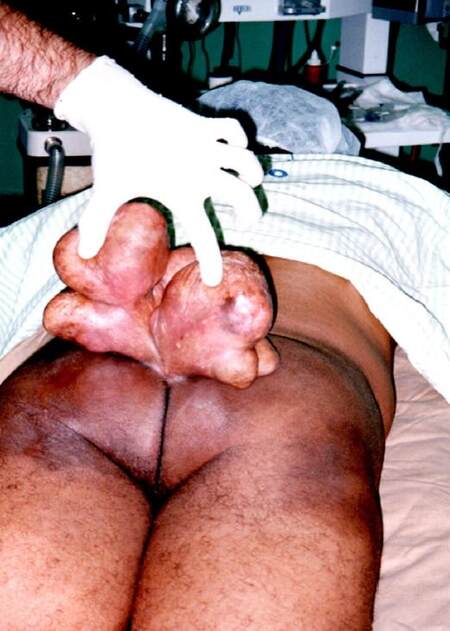
Figure 2. Posterior view of the tumor demonstrating much narrower base that allowed direct excision
Blood tests showed severe anemia (hemoglobin 6.9 g/dL). Computed tomography scan of thoracic and abdominal areas didn’t reveal any tumors or abnormalities with the patient.
The patient was diagnosed with neurofibromatosis, the anemia was corrected with blood transfusion and the patient was taken to the operating room.
Through a direct elipse incision the tumor was dissecated, appearing to be limited to the epidermic region [Figure 3].
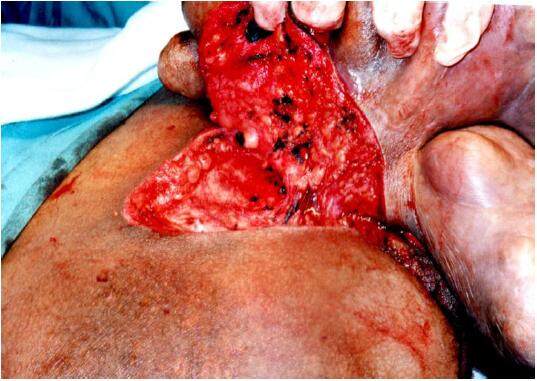
Figure 3. Dissection of the tumor showing limitation to the epidermis
While incising its base, we were not able detect any infiltration on the subjacent subcutaneous tissues. The tumor was completely removed and the surgery was completed with a simple closure of the lateral skin flaps [Figure 4]. The tumor weight was 5,276 g [Figure 5]. The patient was discharged 48 h later after an uneventful postoperative recovery. Histopathological analysis confirmed diagnosis of benign neurofibroma.
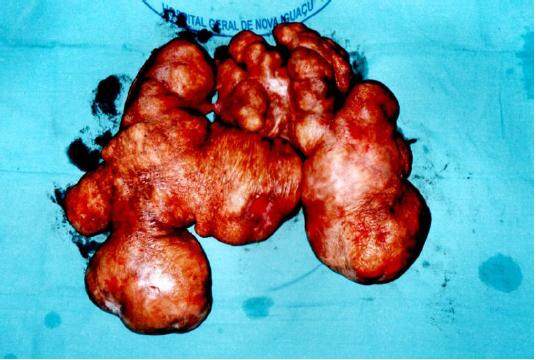
Figure 4. Final aspect of the mass after removal with over 5 kg total weight
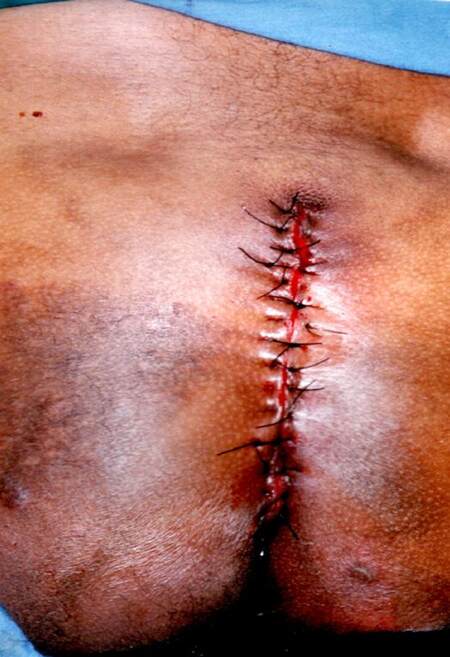
Figure 5. Final aspect of the wound closed
Discussion
Neurofibromas are tumors composed of schwann cells, fibroblasts, mast cells, and vascular components. They can be found in nerves, independently of the location in its surface. Moreover, many authors consider neurofibromas the commonest benign tumors of the NF-1.[3] Neurofibromas can be presented in three subtypes: cutaneous, subcutaneous, and plexiform. Cutaneous lesions and subcutaneous lesions are circumscribed, however none of them have compatible features with NF-1.[4]
During physical examination, neurofibromas may present a pathognomonic buttonhole invagination when pressed with a finger. However, their morphology is not always consistent, resulting in lesions either soft or firm to the touch. Plexiform neurofibromas are noncircumscribed, thick and irregular. This specific subtype for NF-1 can be associated with aesthetic deformation by entwining important supportive structures.[5]
One of the main physical signs encountered along with neurofibromatosis diagnosis is the presence of café au lait spots. These rather unfamiliar pigmentary patterns are brown macules irregularly shaped and evenly pigmented. Usually, when neurofibromatosis is diagnosed, subjects have 6 or more spots that are 1.5 cm or greater in diameter. However, in young children, 5 or more café au lait macules greater than 0.5 cm in diameter are indicative of neurofibromatosis and for which investigation is required. Less than 1% of healthy children have 3 or more of such spots, although 1 or 2 café au lait macules are commonly encountered in healthy individuals without disease. Due to the large volume of the mass presented in this case, it was difficult to determine if the alterations on the skin colour were café au lait spots or decurrent from years of scarification under clothes.[6]
Neurofibromatosis is a dominant autosomal neurogenetic disorder that tends to change and develop with time.[7] Many authors demonstrated the development of this disease with increment of the nerve growth stimulation.[8,9] NF-1 is a pathology that can be associated with different phenotypical manifestations where a group of patients often express cutaneous findings as first symptoms, whilst others might develop life-threatening or severely disfiguring complications. Moreover, amongst individuals from the same family, neurofibromatosis presents itself in different degrees of severity and incidence. The spontaneous mutation rate is 100 times greater than many other genes, and it is considered to contribute to approximately 30-50% of neurofibromatosis cases, however many different mutations in the neurofibromatosis gene have been described.[10]
NF-I is linked to a large gene on band 17q11.2. It encodes a protein called neurofibromin, which has a guanosine triphosphatase region that binds to Ras and positively modulates conversion of guanosine triphosphate to guanosine diphosphate. Several studies confirmed the negative regulation of Ras by this protein, allowing authors to infer that the neurofibromin may act as a tumor suppressor.[11]
Many individuals with this pathology are known to present below average intelligence and some types of learning disabilities, which may comprise neuromotor dysfunction, attention deficit hyperactivity disorder and deficits in visual-spatial processing.
It was also found that 25-40% of individuals with NF-1 might present learning disabilities and up to 5-10% have mental retardation.[4]
It should also be noted that when the lesion has a benign aspect and there are no other signs of malignancy, the excisional biopsia is the treatment of choice and it is normally curative.
Financial support and sponsorship
Nil.
Conflict of interest
There are no conflicts of interest.
REFERENCES
1. Fitzpatrick TB. Neurofibromatosis. In: Color Atlas and Synopsis of Clinical Dermatology. New York, NY: McGraw-Hill; 1997. pp. 458-62.
2. Parry DM, Eldridge R, Kaiser-Kupfer MI, Bouzas EA, Pikus A, Patronas N. Neurofibromatosis 2 (NF2): clinical characteristics of 63 affected individuals and clinical evidence for heterogeneity. Am J Med Genet 1994;52:450-61.
3. Morse RP. Neurofibromatosis type 1. Arch Neurol 1999;56:364-5.
4. National Institutes of Health. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988;45:575-8.
5. Reith JD, Goldblum JR. Multiple cutaneous plexiform schwannomas. Report of a case and review of the literature with particular reference to the association with types 1 and 2 neurofibromatosis and schwannomatosis. Arch Pathol Lab Med 1996;120:399-401.
7. Gutmann DH. Recent insights into neurofibromatosis type 1: clear genetic progress. Arch Neurol 1998;55:778-80.
8. Howell SJ, Wilton P, Lindberg A, Shalet SM. Growth hormone replacement and the risk of malignancy in children with neurofibromatosis. J Pediatr 1998;133:201-5.
9. Brockes JP, Breakefield XO, Martuza RL. Glial growth factor-like activity in Schwann cell tumors. Ann Neurol 1986;20:317-22.
10. Barker D, Wright E, Nguyen K, Cannon L, Fain P, Goldgar D, Bishop DT, Carey J, Baty B, Kivlin J, Willard H, Wayne JS, Greig G, Leinwand L, Nakamura Y, O'Connel P, Leppert M, Lalouel J-M, White R, Skolnick M. Gene for von Recklinghausen neurofibromatosis is in the pericentromeric region of chromosome 17. Science 1987;236:1100-2.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Sforza M, Zaccheddu R, Cancado BL, Sforza D. A case of 25-year-old giant neurofibromatosis. Plast Aesthet Res 2016;3:279-81. http://dx.doi.org/10.20517/2347-9264.2016.46
AMA Style
Sforza M, Zaccheddu R, Cancado BL, Sforza D. A case of 25-year-old giant neurofibromatosis. Plastic and Aesthetic Research. 2016; 3: 279-81. http://dx.doi.org/10.20517/2347-9264.2016.46
Chicago/Turabian Style
Sforza, Marcos, Renato Zaccheddu, Bruno Lopes Cancado, Deborah Sforza. 2016. "A case of 25-year-old giant neurofibromatosis" Plastic and Aesthetic Research. 3: 279-81. http://dx.doi.org/10.20517/2347-9264.2016.46
ACS Style
Sforza, M.; Zaccheddu R.; Cancado BL.; Sforza D. A case of 25-year-old giant neurofibromatosis. Plast. Aesthet. Res. 2016, 3, 279-81. http://dx.doi.org/10.20517/2347-9264.2016.46
About This Article
Copyright

Data & Comments
Data
0

 Cite This Article 3 clicks
Cite This Article 3 clicks

 Like This Article 0
likes
Like This Article 0
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.