
Current concepts in the physiology of adult wound healing

Abstract
Wound healing requires a complex interaction and coordination of different cells and molecules. Any alteration in these highly coordinated events can lead to either delayed or excessive healing. This review provides an overview of adult wound healing physiology. A review of the literature focused on wound healing physiology and current advances in wound healing was conducted using the online Medline/PubMed database. The aim of this review was to inspire further investigation into wound healing physiology that will ultimately translate into improved patient care.
Keywords
Introduction
Wound healing without complications is critical to the survival, as it restores the integrity of the skin and protects the individual from infection and dehydration. Adult wound healing involves a well-orchestrated series of events leading to the repair of injured tissues, resulting in scar formation. Healing of acute wounds, triggered by tissue injury, consists of overlapping and highly coordinated phases of hemostasis, inflammation, proliferation, and remodeling. When a breach of the skin’s integrity occurs, hemostasis is initiated by platelets through fibrin clot formation. Platelets also release various mediators of wound healing to attract macrophages and fibroblasts to the site of tissue injury.[1] The inflammatory phase begins with the arrival of neutrophils followed later by macrophages and lymphocytes at the wound site. The proliferative phase is characterized by new blood vessel formation (angiogenesis), synthesis of extracellular matrix (ECM) components and re-epithelialization.[2] Following the proliferative phase, collagen remodeling begins, along with vascular maturity and regression; this process typically lasts 6-24 months from the time of injury[1][Figure 1].
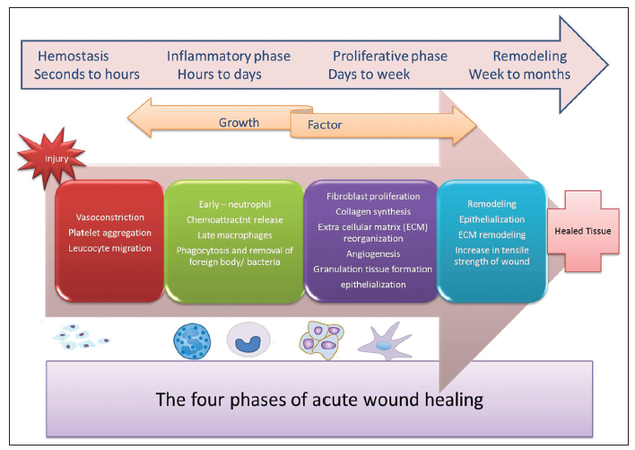
Figure 1. Distinct and overlapping phases of wound healing
The wound healing cascade may be arrested in any of these phases, leading to the formation of a chronic nonhealing wound. Many mediators including inflammatory cells, growth factors, proteases such as matrix metalloproteinase (MMPs) and cellular and extracellular elements play important roles in the process of wound healing. Alterations in one or more of these components may leads to the impaired healing.[2] Wound healing can also be negatively influenced by many exogenous factors, including concurrent disease, such as diabetes, renal failure, malnutrition, smoking, radiation exposure, infection and an immunocompromised state. In the presence of these factors, wounds can fail to heal adequately, resulting in chronic wound formation.[3] The wound healing process can occasionally go into overdrive, resulting in excessive healing and the formation of fibroproliferative scar-like keloids and hypertrophic scars.[4] This review provides a general overview of the physiology of adult wound healing, focusing specifically on how recent advances could translate into improved clinical outcomes.
Hemostasis
Acute wounds cause vascular injury and bleeding from the wound, and the immediate priority is to prevent blood loss by vasoconstriction and formation of a blood clot to seal the vessel. Hemostasis is initiated by the exposure of blood components to the subendothelial layers of the vessel wall. Platelets adhere, aggregate, and form the initial hemostatic plug. The coagulation and complement cascades are then initiated. Within the tissue, prothrombin is activated to form thrombin, which then cleaves fibrinogen to generate fibrin. Along with platelets and the plasma fibronectin, fibrin forms the clot.
The blood clot is made up primarily of cross-linked fibrin, cells such as erythrocytes and platelets, as well as other ECM proteins such as fibronectin, vitronectin and thrombospondin.[5] In addition to containment of blood loss, the blood clot serves as a first defense against microbial invasion and a provisional matrix for the homing of inflammatory cells.[5] The adhesiveness of platelets is mediated by activated integrin receptors on their surface.[6,7] The platelets in the clot undergo degranulation, releasing potent chemoattractants for inflammatory cells, activation factors for local fibroblasts and endothelial cells, and vasoconstrictors, such as chemokine (C-C motif) ligand 5 (CCL5), thrombin, transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), and vascular endothelial growth factor (VEGF).[5] CCL5 is one of the most potent monocyte chemoattractants released by platelets after injury. Thrombin, released by platelets at the wound site, is an early mediator of clot development.[8] Thrombin also induces the release of pro-inflammatory cytokines like CCL2, interleukin-6 (IL-6), and IL‑8 by endothelial cells. These cytokines induce monocyte chemotaxis.[9] Of the injury response chemokines, chemokine (C-X-C motif) ligand 4 (CXCL4) participates in the coagulation process and prevents the premature development of blood vessels.[10] The degradation of fibrin and subsequent activation of the complement system plays a crucial role in mounting the inflammatory process, as well as in facilitating wound angiogenesis and stromal cell proliferation. Fibrin binds to integrin Adhesion molecule CD11b/CD18 on infiltrating monocytes and neutrophils. It also binds to fibroblast growth factor-2 (FGF-2) and VEGF that help the wound tissue vascularize. In addition, fibrin binds to insulin-like growth factor-1 (IGF-1) and promotes stromal cell proliferation.[5,11,12] Under thrombocytopenic conditions, macrophages and T cells at the wound site compensate for the lack of PDGFs and initiation of the inflammatory phase.[13]
Inflammation
The inflammatory process involves the recruitment of neutrophils, macrophages, and lymphocytes. After hemostasis, local vessels dilate secondarily to the effects of the coagulation and complement cascades. Bradykinin (generated by the coagulation cascade) and C3a and C5a anaphylatoxins (generated by the complement cascade) increase blood vessel permeability and attract neutrophils and monocytes to the wound.[14] The C3a and C5a anaphylatoxins also stimulate the release of histamine and leukotrienes from mast cells. The local endothelial cells then break cell-to-cell contact and increase permeability, enhancing the margination of inflammatory cells at the wound site.[15] The initial population of white blood cells in the wound is composed of neutrophils. Thrombin and IL-8 stimulate endothelial permeability through the modulations of adherin-junction endothelial cell adhesion and cell contraction, thereby facilitating leukocyte exit from the circulation.[16,17] Within the wound, neutrophils employ various strategies to kill bacteria and decontaminate the wound, including the secretion of proteases and antimicrobial peptides, as well as the generation of reactive oxygen intermediates via the respiratory burst.[18] In the absence of inflammatory mediators, neutrophils will undergo spontaneous apoptosis. The apoptosis is mediated by cathepsin D release from neutrophil granules, which then facilitates the cleavage and activation of caspase 8, ultimately resulting in caspase 3 activation, DNA fragmentation and apoptosis.[19] In the absence of neutrophils, wound site macrophages lack guidance in conducting the healing process.[20] Although neutrophils play a role in decreasing infection during wound healing, their absence does not prevent the overall progress of wound healing.[21] However, their prolonged presence in the wound may be a factor in the conversion of acute wounds into non-healing chronic wounds.
Within two to three days, monocytes become the predominant inflammatory cell population in the wound. Monocyte chemotaxis to the wound occurs via CC chemokines like CCL2. The chemokines can be released by neutrophils, by the monocytes themselves and by keratinocytes at different stages of healing.[22-24] Circulating monocytes and mast cells are attracted to and infiltrate the wound site.[1,25] Within the wound, monocytes differentiate into macrophages. Macrophages in turn remove apoptotic neutrophils and other dead cells, function as antigen-presenting cells, and secrete cytokines and multiple peptide growth factors.[10] Phagocytosis of the apoptotic neutrophils by macrophages then leads to removal of chemokines from the area of inflammation, preventing further leukocyte influx.[10] Several cytokines and growth factors are known to be secreted by macrophages.[26] Such growth factors include TGF-β, TGF-α, basic FGF (bFGF), VEGF and PDGF. These growth factors activate and attract local endothelial cells, fibroblasts and keratinocytes, and enable wound healing by causing cell proliferation and synthesis of ECM and inducing angiogenesis VEGF,[27-30] which stimulates angiogenesis, also stimulates the macrophages to express LIGHT, a member of the tumor necrosis factor alpha (TNF-α) family of cytokines, which binds to Lymphotoxin-b receptor and induces macrophage death.[31]
Macrophages play a crucial role in enabling wound healing. Macrophage depletion is known to markedly impair wound closure.[27,32] In a landmark study, Leibovich and Ross[33] demonstrated that the antimacrophage serum combined with hydrocortisone diminished the accumulation of macrophages in healing skin wounds of adult guinea pigs. Such depletion resulted in impaired disposal of damaged tissue and provisional matrix, compromised fibroblast count, and delayed healing.[33] Inflammatory responses elicited by injury are only helpful to the healing process if they are timely and transient. However, the inflammation is not essential for skin wound healing. Martin et al.[34] has shown that the PU.1 null mouse, which is devoid of both macrophages and neutrophils, healed both incisional and excisional wounds at statistically similar rates to wild-type littermates, but without scar formation. The cytokine and growth-factor profiles at the wound site in the PU.1 null mouse differed from those of the wild-type. As a result, cell death was reduced, and scar formation did not occur.[34] Studies have focused on platelets and mast cells as targets, and have shown that neither of these mediators is essential to effective wound repair. This further suggests that a dampened or modified inflammatory response could reduce scar formation.[13,35] Impairment of macrophage function at the wound site derails the resolution of inflammation. A persistent inflammatory state of diabetic wound macrophages is caused by impairment in the ability of these cells to phagocytose apoptotic cells at the wound site, in turn preventing the switch from M1 to M2 phenotype.[36] Prolonged inflammation may not only compromise wound closure but may also worsen scar outcomes.[37,38] Lipid mediators, such as the lipoxins, resolvins, protectins, and maresins, have emerged as a novel genus of potent and stereoselective players that counter regulate excessive acute inflammation and stimulate molecular and cellular events that define resolution.[39] The production and activity of several proteases including metalloproteinase, serine proteases and neutrophil elastases which are tightly regulated in acute wound healing may be altered in chronic wounds.[1] For example, non-healing human wound fluid and tissue have increased protease activity, which rapidly degrades exogenously applied peptide growth factors.[40,41] Products targeting excessive protease activity such as protease-scavenging matrices (e.g. Promogran), selective inhibitors, or specific antibodies may be useful in the treatment of chronic wounds refractory to conventional treatments.[42,43]
The lymphocytes are the last type of leukocytes to arrive at the wound site. The lymphocytes exert a specific response against microbes and other foreign material in the wound: B-lymphocytes via antibodies and the T-lymphocytes through production of cytokines and stimulation of cytolytic activity. Lymphocyte-induced inflammation is then resolved by apoptosis when interferon (IFN)‑c and TNF-α are produced at the wound site.[10] Mast cells also appear during the later part of the inflammatory phase, but their function remains unclear. Impaired wound healing has been reported in mast cell-deficient mice.[44] Mast cells have also been implicated in skin wound fibrosis.[45,46] Recently, the role of mast cells in wound healing has become an area of intense research because of a correlation between mast cells and both keloids and hypertrophic scars.[45,46]
Proliferation phase
The proliferative phase of wound healing is accepted to start around two days after injury and typically lasts up to three weeks in a healing cutaneous wound. This phase overlaps with the inflammatory phase, beginning with the degradation of the initial fibrin-platelet matrix and invasion of fibroblasts and endothelial cells. Proteases of the serine, cysteine, and MMPs families are secreted to facilitate cellular migration through the fibrin clot and provisional matrix.[47-50] The major events of this phase include the influx of fibroblasts, ECM deposition, formation of new blood vessels, and re-epithelialization.
Fibroblasts are the key type of cells in this phase of healing and become the predominant cell type by three to five days after injury. Macrophages and mast cells release growth factors, including PDGF and TGF-β, that stimulate fibroblast activation.[25] The fibroblasts proliferate and produce the matrix proteins fibronectin, hyaluronic acid, collagen and proteoglycans, all of which help to construct the new ECM and a platform for keratinocyte migration.[1,14] The provisional fibrin matrix is gradually replaced by granulation tissue.
Granulation tissue is a dense conglomeration of blood vessels, macrophages and fibroblasts embedded within a loose matrix of fibronectin, hyaluronic acid, and collagen. Granulation tissue begins to appear in human wounds by about four days after injury. During granulation tissue formation, new blood vessels develop from preexisting vessels (angiogenesis). Angiogenic factors are secreted by fibroblasts and macrophages (e.g. VEGF, basic FGF, angiopoietin1 and thrombospondin), keratinocytes (e.g. CXCL8 and VEGF), and endothelial cells themselves (e.g. CXCL8 and VEGF).[51-54] Integrin avb3 at the leading capillary tipis a prerequisite for endothelial growth, and is a promising therapeutic target for angiogenesis.[55] Blocking these processes with angiogenesis inhibitors impairs wound healing and can be corrected with growth factors such as VEGF.[51] Over time, the fibrin provisional matrix is replaced with type III collagen, which in turn is replaced by type I collagen during the remodeling phase. At least twenty‑eight different types of collagen are currently known.[56] Most collagen types in the ECM are synthesized by fibroblasts; however, some types are synthesized by keratinocytes.[57]
Approximately four days after injury, myofibroblasts appear in the wound.[58] TGF-β and CXCL8 promote the differentiation of fibroblasts in the granulation tissue into myofibroblasts.[48,59,60] Myofibroblast differentiation also requires an interaction with cellular fibronectin containing the extra domain‑A domain. Inhibition of either fibronectin or the corresponding integrin receptors prevents TGF-β1-mediated myofibroblast differentiation.[61,62] Myofibroblasts exert their contractile forces by focal adhesion contacts that link the intracellular cytoskeleton to the ECM. In vitro experiments have shown higher contractile forces of keratinocytes compared with fibroblasts.[63]
Re-epithelialization is an important process during wound healing that starts in the early phase of healing. Platelets in the early wound release epidermal growth factor (EGF) and TGF-β, stimulate the keratinocytes at the wound edge to proliferate and migrate to cover the wound. Cytokines PDGF, TNF-α, FGF, keratinocyte growth factor and CXCL8 produced by neutrophils, macrophages, endothelial cells and fibroblasts, maintain the proliferation and migration of keratinocytes which in turn induces wound re-epithelialization.[22,64-66] During re-epithelialization, the keratinocytes migrate beneath the provisional ECM. MMP release keratinocytes from their substratum and help in the migration through the matrix and promotion of re-epithelialization.[67-69] Wound treatment with a broad-spectrum metalloproteinase inhibitor significantly delays re-epithelialization in vitro and in vivo.[70,71] Wound re-epithelialization also requires the activity of various proteases, including the serine protease plasmin. Re-epithelialization is delayed in plasminogen-deficient mice, due to the inability of keratinocytes to degrade and thus migrate through the fibrin matrix and the underlying dermal tissue, whereas mice deficient in both plasminogen and fibrinogen exhibit more normal healing.[72,73] After the re-establishment of the epithelial layer, keratinocytes and fibroblasts secrete type IV collagen to form the basement membrane.[74] The keratinocytes undergo division and become columnar to restore the epidermal layer and reform a barrier to infection and moisture loss.
The dysregulation of the proliferative phase is believed to underlie the pathophysiology of chronic wound and fibrotic disorders such as hypertrophic scarring and keloids. A randomized controlled trial in patients with diabetic neuropathic foot ulcers showed topical PDGF to be superior to placebo in promoting healing.[75] VEGF gene transfer was effective in increasing vascularity in ischemic leg ulcers.[76] Among cytokines and growth factors, the possible targets for promotion of wound healing include TNF-α, PDGF, FGF, VEGF, IGF-1, and EGF.[77-80] Understanding the signals for halting the proliferative phase will help developing new therapeutics for acute wound healing.[51]
Remodeling phase
The remodeling of wound tissue occurs over a prolonged time and may last up to 1 year.[51] It involves ECM turnover coupled with a significant decrease in cellularity. The decline in cellularity results from the apoptosis of residual inflammatory cells and myofibroblasts as well as regression of the neovasculature.[59] In humans, remodeling is characterized by both wound contraction and collagen remodeling. The balance of collagen metabolism is in part determined by the regulation of MMP activity.[81] The process of wound contraction is produced by wound myofibroblasts. While remodeling, wounds gradually become stronger with time. Wound tensile strength increases rapidly from 1 to 8 weeks after wounding and correlates with collagen cross-linking by lysyl oxidase.[82] The tensile strength of wounded skin reaches at best only approximately 80% that of unwounded skin, but can be increased by synthetic MMPs inhibitors.[83,84] Scar formation is the final outcome of wound repair in children and adults.
New therapeutic strategies can be tried to reduce an aesthetically unacceptable scar appearance. Treatment with TGF-β3 formulations and neutralizing antibodies to TGF-β as well as solutions that decrease the activity of connexin 43, a mediator of TGF-β signaling, has been shown to reduce the inflammatory response and scar formation.[85,86] Evidence of success using TGF-β-related strategies was provided by a study showing that the exogenous addition to wounds of fibromodulin, a TGF-β modulator, reduces scar.[87] Decorin is a small chondroitin/dermatan sulfate proteoglycan that limits the duration of TGF-β influence on inflammation and tissue repair, promoting regenerative repair and limiting tissue fibrosis.[88] Other novel strategies include the application of antifibrotic human recombinant growth factors and cytokines, anti-inflammatory substances, protease inhibitors and molecules that interfere with profibrotic cytokine function (e.g. TGF-β) and collagen synthesis at the wound site.[89]
Fetal wound healing
Fetal wound healing is an area of great interest because it is characterized by scar-less, regenerative wound healing. This process is age-dependent, like postnatal healing, wounds in third-trimester cause scarring.[90] The exact mechanism responsible for scar-less healing in the first and second trimesters is not yet clearly understood. The proposed mechanisms include decreased inflammation, unique properties of fetal cells, altered cytokine milieu, variable gene expression and ECM deposition.[91] Recent fields of research revolve around the role of TGF, IL-10 and mast cells. Alice et al.[92] described a major role for IL-10 in scar-less wound healing. The authors propose a "cytokine hypothesis" centered on the anti-inflammatory properties of IL-10. IL-10 protects against excess deposition of collagen, maintains elevated hyaluronic levels, enhances fibroblast function, prevents differentiation of fibroblast to myofibroblasts and increases survival of endothelial progenitor cells and angiogenesis.[92] Research in a mouse model demonstrated the scarring potential of mast cells in fetal wounds. In early fetal life (day 15), scar-less wounds were associated with a lesser number of mast cells with reduced degranulation as compared to later scarring wounds.[93] Another factor implicated in fetal wound healing is the growth factor TGF-β. Of the three isoforms, TGF-β1 is responsible for fibrosis. TGF-β3 isoform is the predominant isotype in fetal wound healing, and altered profiling of the isoform may be factor responsible for scar-less healing. Other additional mechanisms include mediators of TGF pathway such as connective tissue growth factor, proteoglycan, decorin and P311.[94]
Role of stem cells in wound healing
Stem cells are a specialized group of cells with the potential for self-renewal, as well as the ability to differentiate into various cell lineages. Stem cells can be classified according to their origin (embryonic, fetal and adult) or based on the differentiation potential (totipotent, pluripotent, multipotent and unipotent).[95] Due to the ease of availability and fewer ethical issues, adult stem cells are the most commonly used type of stem cells in medical practice.
Mesenchymal stem cells are derived from bone marrow, adipose tissue, umbilical cord, periosteum, tendons, muscle and skin.[96] The most commonly used source is adipose tissue. Stem cells affect all stages of wound healing. They have significant anti-inflammatory and immunomodulatory effects in the inflammation phase of healing.[97] In the proliferative phase they also stimulate fibroblasts, keratinocytes and endothelial cells, thereby accelerating wound closure. Uysal et al.[98] demonstrated that wound healing time was reduced in rats treated by patchy skin grafts and mesenchymal stem cells. In addition, wound contraction was reduced, angiogenesis was increased, epithelization progressed rapidly.[99]
Stem cell therapy can be administered either topically or systemically. Falanga et al.[100] demonstrated a topical application of mesenchymal stem cells with either fibrinogen or thrombin applied to chronic wounds in the form of a spray. This spray is converted into a gel form over the wound and helps in retaining the stem cells over the wound.[100] To improve the retention of stem cells in the wound, cells are now applied on an adequate support/scaffold like collagen, skin substitutes. This helps in maintaining the viability of the cells and facilitates migration in the wound bed.[101]
Conclusion
Cutaneous wound healing is a complex and dynamic biological process requiring the interaction and coordination of many different cell types and molecules, including growth factors and cytokines. Tremendous strides have been made in delineating the myriad of factors involved in normal and delayed/excessive healing. However, this increased understanding has not led to significant advances in patient care. Administration of exogenous growth factors and cytokines has shown promise in improving healing results in wounds. As wound healing involves multiple molecular mechanisms, no single agent therapy is likely to be successful in accelerating or modulating wound healing.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
4. Köse O, Waseem A. Keloids and hypertrophic scars: are they two different sides of the same coin? Dermatol Surg 2008;34:336-46.
5. SenCK, Roy S. Wound healing. In: Rodriguez E, Losee J, Neligan PC, editors. Plastic Surgery: Volume 3: Craniofacial, Head and Neck Surgery and Pediatric Plastic Surgery. 3rd ed. Philadelphia: Saunders; 2012. pp. 240-66.
6. Bennett JS, Berger BW, Billings PC. The structure and function of platelet integrins. J Thromb Haemost 2009;7:200-5.
7. Suzuki-Inoue K. Activation and inhibitory mechanisms of blood platelets. Nihon Rinsho 2014;72:1212-7.
8. He S, Blombäck M, Bark N, Johnsson H, Wallén NH. The direct thrombin inhibitors (argatroban, bivalirudin and lepirudin) and the indirect Xa-inhibitor (danaparoid) increase fibrin network porosity and thus facilitate fibrinolysis. Thromb Haemost 2010;103:1076-84.
9. Marin V, Montero-Julian FA, Grès S, Boulay V, Bongrand P, Farnarier C, Kaplanski G. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: an experimental model involving thrombin. J Immunol 2001;167:3435-42.
10. Martins-Green M, Petreaca M, Wang L. Chemokines and their receptors are key players in the orchestra that regulates wound healing. Adv Wound Care 2013;2:327-47.
11. Sahni A, Odrljin T, Francis CW. Binding of basic fibroblast growth factor to fibrinogen and fibrin. J Biol Chem 1998;273:7554-9.
12. Tuan TL, Wu H, Huang EY, Chong SS, Laug W, Messadi D, Kelly P, Le A. Increased plasminogen activator inhibitor-1 in keloid fibroblasts may account for their elevated collagen accumulation in fibrin gel cultures. Am J Pathol 2003;162:1579-89.
13. Szpaderska AM, Egozi EI, Gamelli RL, DiPietro LA. The effect of thrombocytopenia on dermal wound healing. J Invest Dermatol 2003;120:1130-7.
14. Mirastschijski U, Jokuszies A, Vogt PM. Skin wound healing: repair biology, wound and scar treatment. In: Rodriguez E, Losee J, Neligan PC, editors. Plastic Surgery: Volume 3: Craniofacial, Head and Neck Surgery and Pediatric Plastic Surgery. 3rd ed. Philadelphia: Elsevier; 2012. pp. 268-96.
15. Roberts HR, Tabares AH. Overview of the coagulation reactions. In: High KA, Roberts HR, editors. Molecular basis of thrombosis and hemostasis. New York: Marcel Dekker; 1995. pp. 35-50.
16. Petreaca ML, Yao M, Liu Y, Defea K, Martins-Green M. Transactivation of vascular endothelial growth factor receptor-2 (VEGFR2) by interleukin-8 (IL-8/CXCL8) is required for IL-8/CXCL8-induced endothelial permeability. MolBiol Cell 2007;18:5014-23.
17. Schraufstatter IU, Chung J, Burger M. IL-8 activates endothelial cell CXCR1 and CXCR2 through Rho and Rac signaling pathways. Am J Physiol Lung Cell Mol Physiol 2001;280:1094-103.
18. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006;6:173-82.
19. Conus S, Perozzo R, Reinheckel T, Peters C, Scapozza L, Yousefi S, Simon HU. Caspase-8 is activated by cathepsin D initiating neutrophil apoptosis during the resolution of inflammation. J Exp Med 2008;205:685-98.
20. Peters T, Sindrilaru A, HinzB, Hinrichs R, Menke A, Al-Azzeh EA, Holzwarth K, OreshkovaT, Wang H, Kess D, Walzog B, Sulyok S, Sunderkötter C, Friedrich W, Wlaschek M, Krieg T, Scharffetter-Kochanek K. Wound-healing defect of CD18 (-/-) mice due to a decrease in TGF-beta1 and myofibroblast differentiation. EMBO J 2005;24:3400-10.
21. Simpson DM, Ross R. The neutrophilic leukocyte in wound repair: a study with antineutrophil serum. J Clin Invest 1972;51:2009-23.
23. DiPietro LA, Polverini PJ, Rahbe SM, Kovacs EJ. Modulation of JE/MCP-1 expression in dermal wound repair. Am J Pathol 1995;146:868-75.
24. Wetzler C, Kampfer H, Pfeilschifter J, Frank S. Keratinocyte-derived chemotactic cytokines: expressional modulation by nitric oxide in vitro and during cutaneous wound repair in vivo. Biochem Biophys Res Commun 2000;274:689-96.
25. Martin P, Leibovich SJ. Inflammatory cells during wound repair: the good, the bad and the ugly. Trends Cell Biol 2005;15:599-607.
26. Novak ML, Koh TJ. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol 2013;183:1352-63.
27. Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol 2007;127:514-25.
28. Eming SA, Werner S, Bugnon P, Wickenhauser C, Siewe L, Utermöhlen O, Davidson JM, Krieg T, Roers A. Accelerated wound closure in mice deficient for interleukin-10. Am J Pathol 2007;170:188-202.
29. Miao M, Yuan B, Mani R, Lu S. Macrophage activation dysfunction in impaired wound healing: a potential therapeutic target. Int J Low Extrem Wounds 2013;12:239-41.
30. Sindrilaru A, Scharffetter-Kochanek K. Disclosure of the culprits: macrophages-versatile regulators of wound healing. Adv Wound Care (New Rochelle) 2013;2:357-68.
31. Petreaca ML, Yao M, Ware C, and Martins-Green MM. Vascular endothelial growth factor promotes macrophage apoptosis through stimulation of tumor necrosis factor superfamily member 14 (TNFSF14/LIGHT). Wound Repair Regen 2008;16:602-14.
32. DiPietro LA. Wound healing: the role of the macrophage and other immune cells. Shock 1995;4:233-40.
33. Leibovich SJ, Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol 1975;78:71-100.
34. Martin P, D'Souza D, Martin J, Grose R, Cooper L, Maki R, McKercher SR. Wound healing in the PU.1 null mouse-tissue repair is not dependent on inflammatory cells. Curr Biol 2003;13:1122-8.
35. Egozi EI, Ferreira AM, Burns AL, Gamelli RL, Dipietro LA. Mast cells modulate the inflammatory but not the proliferative response in healing wounds. Wound Repair Regen 2003;11:46-54.
36. Khanna S, Biswas S, Shang Y, Collard E, Azad A, KauhC, Bhasker V, Gordillo GM, Sen CK, Roy S. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 2010;5:e9539.
37. Gordon A, Kozin ED, Keswani SG, Vaikunth SS, Katz AB, Zoltick PW, Favata M, Radu AP, Soslowsky LJ, Herlyn M, Crombleholme TM. Permissive environment in postnatal wounds induced by adenoviral-mediated overexpression of the anti-inflammatory cytokine interleukin-10 prevents scar formation. Wound Repair Regen 2008;16:70-9.
38. Li P, Liu P, Xiong RP, Chen XY, Zhao Y, Lu WP, Liu X, Ning YL, Yang N, Zhou YG. Ski, a modulator of wound healing and scar formation in the rat skin and rabbit ear. J Pathol 2011;223:659-71.
39. Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ Res 2010;107:1170-84.
40. Mirastschijski U, Impola U, Jahkola T, Karlsmark T, AGren MS, Saarialho-Kere U. Ectopic localization of matrix metalloproteinase-9 in chronic cutaneous wounds. Hum Pathol 2002;33:355-64.
41. Yager DR, Nwomeh BC. The proteolytic environment of chronic wounds. Wound Repair Regen 1999;7:433-41.
42. Enoch S, Grey JE, Harding KG. ABC of wound healing. Non-surgical and drug treatments. BMJ 2006;332:900-3.
43. Fray MJ, Dickinson RP, Huggins JP, Occleston NL. A potent, selective inhibitor of matrix metalloproteinase-3 for the topical treatment of chronic dermal ulcers. J Med Chem 2003;46:3514-25.
44. Shiota N, Nishikori Y, Kakizoe E, Shimoura K, Niibayashi T, ShimboriC, Tanaka T, Okunishi H. Pathophysiological role of skin mast cells in wound healing after scald injury: study with mast cell-deficient W/W (V) mice. Int Arch Allergy Immunol 2010;151:80-8.
45. Gallant-Behm CL, Hildebrand KA, Hart DA. The mast cell stabilizer ketotifen prevents development of excessive skin wound contraction and fibrosis in red Duroc pigs. Wound Repair Regen 2008;16:226-33.
47. Lund LR, Rømer J, Bugge TH, Nielsen BS, Frandsen TL, DegenJL, Stephens RW, Danø K. Functional overlap between two classes of matrix-degrading proteases in wound healing. EMBO J 1999;18:4645-56.
48. Mirastschijski U, Schnabel R, Claes J, Schneider W, Agren MS, Haaksma C, Tomasek JJ. Matrix metalloproteinase inhibition delays wound healing and blocks latent transforming growth factor-β1 promoted myofibroblast formation and function. Wound Repair Regen 2010;18:223-34.
49. Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol 2001;17:463-516.
50. Brix K, Dunkhorst A, Mayer K, Jordans S. Cysteine cathepsins: cellular roadmap to different functions. Biochimie 2008;90:194-207.
51. Gurtner GC. Wound healing: normal and abnormal. In: Thorne CH, Bartlett SP, Beasley RW, Aston SJ, Gurtner GC, Spear SL, editors. Grabb and Smith's Plastic Surgery. 6th ed. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 15-22.
52. Johnson KE, Wilgus TA. Vascular Endothelial Growth Factor and Angiogenesis in the Regulation of Cutaneous Wound Repair. Adv Wound Care (New Rochelle) 2014;3:647-61.
53. Cao PF, Xu YB, Tang JM, Yang RH, Liu XS. HOXA9 regulates angiogenesis in human hypertrophic scars: induction of VEGF secretion by epidermal stem cells. Int J Clin Exp Pathol 2014;7:2998-3007.
54. Kazemi-Lomedasht F, Behdani M, Bagheri KP, Habibi-Anbouhi M, Abolhassani M, Arezumand R, Shahbazzadeh D, Mirzahoseini H. Inhibition of angiogenesis in human endothelial cell using VEGF specific nanobody. Mol Immunol 2015;65:58-67.
55. Li YJ, Li XH, Wang LF, Kuang X, Hang ZX, Deng Y, Du JR. Therapeutic efficacy of a novel non-peptide αvβ3 integrin antagonist for pathological retinal angiogenesis in mice. Exp Eye Res 2014;129:119-26.
56. Mienaltowski MJ, Birk DE. Structure, physiology, and biochemistry of collagens. Adv Exp Med Biol 2014;802:5-29.
57. Nyström A, Velati D, Mittapalli VR, Fritsch A, Kern JS, Bruckner‑Tuderman L. Collagen VII plays a dual role in wound healing. J Clin Invest 2013;123:3498-509.
58. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3:349-63.
59. Desmoulière A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen 2005;13:7-12.
60. Feugate JE, Li Q, Wong L, and Martins-Green M. The cxc chemokine cCAF stimulates differentiation of fibroblasts into myofibroblasts and accelerates wound closure. J Cell Biol 2002;156:161-72.
61. Kato R, Kamiya S, Ueki M, Yajima H, Ishii T, Nakamura H, Katayama T, Fukai F. The fibronectin-derived antiadhesive peptides suppress the myofibroblastic conversion of rat hepatic stellate cells. Exp Cell Res 2001;265:54-63.
62. Darby IA, Laverdet B, Bonté F, Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin Cosmet Investig Dermatol 2014;7:301-11.
63. Wall IB, Bhadal N, Broad S, Whawell SA, Mudera V, Lewis MP. Force generation and protease gene expression in organotypic co-cultures of fibroblasts and keratinocytes. J Tissue Eng Regen Med 2009;3:647-50.
64. Shah JM, Omar E, Pai DR, Sood S. Cellular events and biomarkers of wound healing. Indian J Plast Surg 2012;45:220-8.
66. Werner S, Krieg T, Smola H. Keratinocyte-fibroblast interactions in wound healing. J Invest Dermatol 2007;127:998-1008.
67. Fu X, Xu M, Liu J, Qi Y, Li S, Wang H. Regulation of migratory activity of human keratinocytes by topography of multiscale collagen-containing nanofibrous matrices. Biomaterials 2014;35:1496-506.
68. Stevens LJ, Page-McCaw A. A secreted MMP is required for reepithelialization during wound healing. Mol Biol Cell 2012;23:1068-79.
69. Dumin JA, Dickeson SK, Stricker TP, Bhattacharyya-Pakrasi M, Roby JD, Santoro SA, Parks WC. Pro-collagenase-1 (matrix metalloproteinase-1) binds the alpha(2)beta(1) integrin upon release from keratinocytes migrating on type I collagen. J Biol Chem 2001;276:29368-74.
70. Mirastschijski U, Haaksma CJ, Tomasek JJ, Agren MS. Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds. Exp Cell Res 2004;299:465-75.
71. Mirastschijski U, Impola U, Karsdal MA, Saarialho-Kere U, Agren MS. Matrix metalloproteinase inhibitor BB-3103 unlike the serine proteinase inhibitor aprotinin abrogates epidermal healing of human skin wounds ex vivo. J Invest Dermatol 2002;118:55-64.
72. Frøssing S, Rønø B, Hald A, Rømer J, Lund LR. Skin wound healing in MMP2-deficient and MMP2/plasminogen double-deficient mice. Exp Dermatol 2010;19:e234-40.
73. Green KA, Almholt K, Ploug M, Rønø B, Castellino FJ, Johnsen M, Bugge TH, Rømer J, Lund LR. Profibrinolytic effects of metalloproteinases during skin wound healing in the absence of plasminogen. J Invest Dermatol 2008;128:2092-101.
74. Wojtowicz AM, Oliveira S, Carlson MW, Zawadzka A, Rousseau CF, Baksh D. The importance of both fibroblasts and keratinocytes in a bilayered living cellular construct used in wound healing. Wound Repair Regen 2014;22:246-55.
75. Steed DL. Clinical evaluation of recombinant human platelet-derived growth factor for the treatment of lower extremity ulcers. Plast Reconstr Surg 2006;117:S143-9.
76. Mäkinen K, Manninen H, Hedman M, Matsi P, Mussalo H, Alhava E, Ylä-Herttuala S. Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: a randomized, placebo-controlled, double-blinded phase II study. Mol Ther 2002;6:127-33.
77. Greaves NS, Ashcroft KJ, Baguneid M, Bayat A. Current understanding of molecular and cellular mechanisms in fibroplasia and angiogenesis during acute wound healing. J Dermatol Sci 2013;72:206-17.
78. Kang HC Ahn SD, Choi DH, Kang MK, Chung WK, Wu HG. The safety and efficacy of EGF-based cream for the prevention of radiotherapy-induced skin injury: results from a multicenter observational study. Radiat Oncol J 2014;32:156-62.
79. Singla S, Garg R, Kumar A, Gill C. Efficacy of topical application of beta urogastrone (recombinant human epidermal growth factor) in Wagner's Grade 1 and 2 diabetic foot ulcers: Comparative analysis of 50 patients. J Nat Sci Biol Med 2014;5:273-7.
80. Barrientos S, Brem H, Stojadinovic O, Tomic-Canic M. Clinical application of growth factors and cytokines in wound healing. Wound Repair Regen 2014;22:569-78.
81. Toriseva M, Kahari VM. Proteinases in cutaneous wound healing. Cell Mol Life Sci 2009;66:203-24.
82. Lau YK, Gobin AM, West JL. Overexpression of lysyl oxidase to increase matrix crosslinking and improve tissue strength in dermal wound healing. Ann Biomed Eng 2006;34:1239-46.
83. Gurtner GC, Wong VW. Wound healing: normal and abnormal. In: Thorne CH, Gurtner GC, Chung K, Gosain A, Mehrara B, Rubin P, Spear SL, editors. Grabb and Smith's Plastic Surgery. 7th ed. Philadelphia: Lippincott Williams & Wilkins; 2013. pp. 13-9.
84. Levenson SM, Geever EF, Crowley LV, Oates JF 3rd, Berard CW, Rosen H. The healing of rat skin wounds. Ann Surg 1965;161:293-308.
85. Witte MB, Thornton FJ, Kiyama T, Efron DT, Schulz GS, Moldawer LL, Barbul A. Metalloproteinase inhibitors and wound healing: a novel enhancer of wound strength. Surgery 1998;124:464-70.
86. Viera MH, Vivas AC, Berman B. Update on keloid management: clinical and basic science advances. Adv Wound Care (New Rochelle) 2012;1:200-6.
87. Rhett JM, Ghatnekar GS, Palatinus JA, O'Quinn M, Yost MJ, Gourdie RG. Novel therapies for scar reduction and regenerative healing of skin wounds. Trends Biotechnol 2008;26:173-80.
88. Soo C, Hu FY, Zhang X, Wang Y, Beanes SR, Lorenz HP, Hedrick MH, Mackool RJ, Plaas A, Kim SJ, Longaker MT, Freymiller E, Ting K. Differential expression of fibromodulin, a transforming growth factor-beta modulator, in fetal skin development and scarless repair. Am J Pathol 2000;157:423-33.
89. Järveläinen H, Puolakkainen P, Pakkanen S, Brown EL, Höök M, Iozzo RV, Sage EH, Wight TN. A role for decorin in cutaneous wound healing and angiogenesis. Wound Repair Regen 2006;14:443-52.
90. Occleston NL, O'Kane S, Goldspink N, Ferguson MW. New therapeutics for the prevention and reduction of scarring. Drug Discov Today 2008;13:973-81.
91. Wilgus TA. Regenerative healing in fetal skin: a review of the literature. Ostomy Wound Manage 2007;53:16-31.
92. King A, Balaji S, Le LD, Crombleholme TM, Keswani SG. Regenerative wound healing: the role of Interleukin-10. Adv Wound Care (New Rochelle) 2014;3:315-23.
93. Lo DD, Zimmermann AS, Nauta A, Longaker MT, Lorenz HP. Scarless fetal skin wound healing update. Birth Defects Res C Embryo Today 2012;96:237-47.
94. Wulff BC, Parent AE, Meleski MA, DiPietro LA, Schrementi ME, Wilgus TA. Mast cells contribute to scar formation during fetal wound healing. J Invest Dermatol 2012;132:458-65.
95. Penn JW, Grobbelaar AO, Rolfe KJ. The role of the TGF-β family in wound healing, burns and scarring: a review. Int J Burns Trauma 2012;2:18-28.
96. Koźlik M, Wójcicki P. The use of stem cells in plastic and reconstructive surgery. Adv Clin Exp Med 2014;23:1011-17.
97. da Silva Meirelles L, Caplan AI, Nardi NB. In search of the in vivo identity of mesenchymal stem cells. Stem Cells 2008;26:2287-99.
98. Uysal CA, Tobita M, Hyakusoku H, Mizuno H. The Effect of Bone-Marrow-Derived Stem Cells and Adipose-Derived Stem Cells on Wound Contraction and Epithelization. Adv Wound Care (New Rochelle) 2014;3:405-13.
99. Singer NG, Caplan AI. Mesenchymal stem cells: mechanisms of inflammation. Annu Rev Pathol 2011;6:457-78.
100. Falanga V, Iwamoto S, Chartier M, Yufit T, Butmarc J, Kouttab N, Shrayer D, Carson P. Autologous bone marrow-derived cultured mesenchymal stem cells delivered in a fibrin spray accelerate healing in murine and human cutaneous wounds. Tissue Eng 2007;13:1299-312.
Cite This Article
Export citation file: BibTeX | RIS
OAE Style
Thiruvoth FM, Mohapatra DP, Kumar D, Chittoria SRK, Nandhagopal V. Current concepts in the physiology of adult wound healing. Plast Aesthet Res 2015;2:250-6. http://dx.doi.org/10.4103/2347-9264.158851
AMA Style
Thiruvoth FM, Mohapatra DP, Kumar D, Chittoria SRK, Nandhagopal V. Current concepts in the physiology of adult wound healing. Plastic and Aesthetic Research. 2015; 2: 250-6. http://dx.doi.org/10.4103/2347-9264.158851
Chicago/Turabian Style
Thiruvoth, Friji Meethale, Devi Prasad Mohapatra, Dinesh Kumar, Sivakuma Ravi Kumar Chittoria, Vijayaraghavan Nandhagopal. 2015. "Current concepts in the physiology of adult wound healing" Plastic and Aesthetic Research. 2: 250-6. http://dx.doi.org/10.4103/2347-9264.158851
ACS Style
Thiruvoth, FM.; Mohapatra DP.; Kumar D.; Chittoria SRK.; Nandhagopal V. Current concepts in the physiology of adult wound healing. Plast. Aesthet. Res. 2015, 2, 250-6. http://dx.doi.org/10.4103/2347-9264.158851
About This Article
Special Issue
Copyright

Data & Comments
Data


 Cite This Article 244 clicks
Cite This Article 244 clicks

 Like This Article 10
likes
Like This Article 10
likes






Comments
Comments must be written in English. Spam, offensive content, impersonation, and private information will not be permitted. If any comment is reported and identified as inappropriate content by OAE staff, the comment will be removed without notice. If you have any queries or need any help, please contact us at support@oaepublish.com.